Mirrorstoolkit: one-liner analysis tool
Yihao Li
Reyes-Soffer Lab, Division of Preventive medicine, Department of Medicine, Columbia University Irving Medical Center;Badri Vardarajan Lab, Gertrude H. Sergievsky Center, Department of Neurology, Columbia University Irving Medical Centerintroduction.Rmd
library(dplyr)
#> Warning: package 'dplyr' was built under R version 4.4.3
#>
#> Attaching package: 'dplyr'
#> The following objects are masked from 'package:stats':
#>
#> filter, lag
#> The following objects are masked from 'package:base':
#>
#> intersect, setdiff, setequal, union
library(readxl)
library(mirrorstoolkit)Data Preparation
Compute NIH equation 2 for LDL (calc) estimation
compute_nih_ldl functions allows either value or vectorized operation
By value
Simply input the value
mirrorstoolkit::compute_nih_ldl(tchol = 112.9,hdl = 39.9, trig = 151)
#> [1] 47.18626Vectorized
Use the column name, it uses basic dplyr naming flow. If your column name is not one character string surround it by “`”
df = df %>% dplyr::mutate(
`LDL (calc) mg/dL` = mirrorstoolkit::compute_nih_ldl(
tchol = `Chol mg/dL`,
hdl = `HDL mg/dL`,
trig = `Trig mg/dL`))
df %>%
dplyr::select(
tidyr::all_of(
c("Chol mg/dL",
"HDL mg/dL",
"Trig mg/dL",
"LDL (calc) mg/dL")
)) %>% head()
#> # A tibble: 6 × 4
#> `Chol mg/dL` `HDL mg/dL` `Trig mg/dL` `LDL (calc) mg/dL`
#> <dbl> <dbl> <dbl> <dbl>
#> 1 113. 39.9 151 47.2
#> 2 189. 76.5 146 87.8
#> 3 92 49.2 82 26.1
#> 4 153. 29.4 315 73.0
#> 5 134. 58.8 83 59.7
#> 6 105. 41.3 145 38.7Exploratory Data Analysis
Single Group analysis
Normality Inspection
This is a part which I feel is not emphasized enough in our lab practice - we need to determine if a lab evaluation or result result is normally distributed. inspect_distribution() can give a basic visualization about the distribution by
- The histogram
- The density plot
- Shapiro-Wilk test result - please be aware that the null hypothesis is it’s normally distributed, thus if it is significant, it is not normal.
It usually gives a lot of warnings (depends on the completeness and quality of your data for most of the times), I suppressed them here for tidiness, but expose the warnings (omit the last function) are recommended, it will give you useful informations.
Please always be aware that the normality it’s a very subjective decision, always add a huge grab of your own salts from your reading and experience, especially when you have a small sample size. See below, from my perspective:
- Albumin, from PMID: 18405401, is generally normal overall.
- Calcium is quite normal, which is a bit out of my expectation.
- Smoking is actually boolean (binary, 0/1).
- LDL, it is on you, you can either treat it as normal or not normal, my experience is it’s generally normal.
- wIS often treated as normally distributed on my side.
- Lp(a), skewed right for sure
mirrorstoolkit::inspect_distribution(
data = df,
variable = c(
"Albumin",
"Calcium",
"Smoking",
"LDL (calc) mg/dL",
"wIS",
"Lp(a) (nmol/L)")
) %>% base::suppressWarnings()
#> $visualization
#> $`1`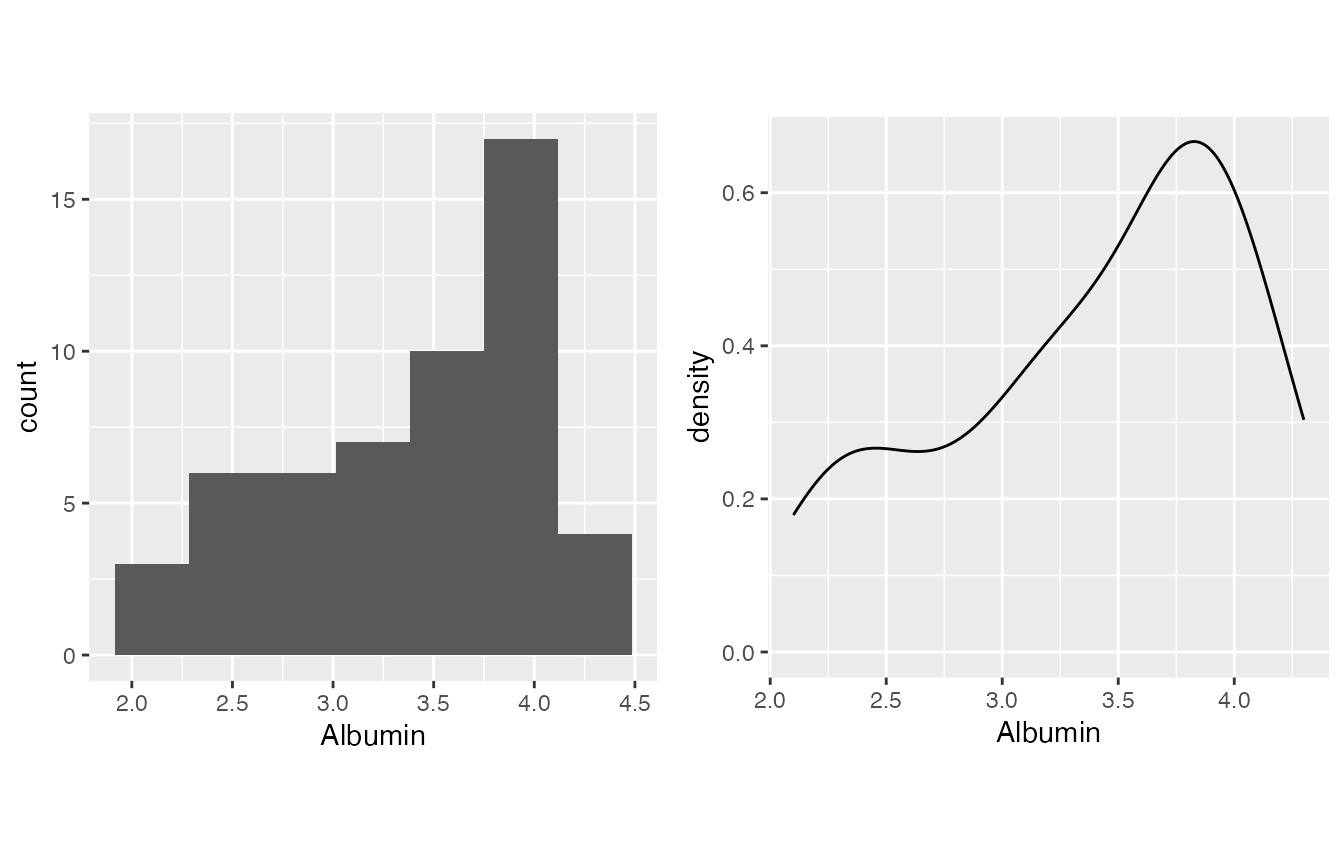
#>
#> $`2`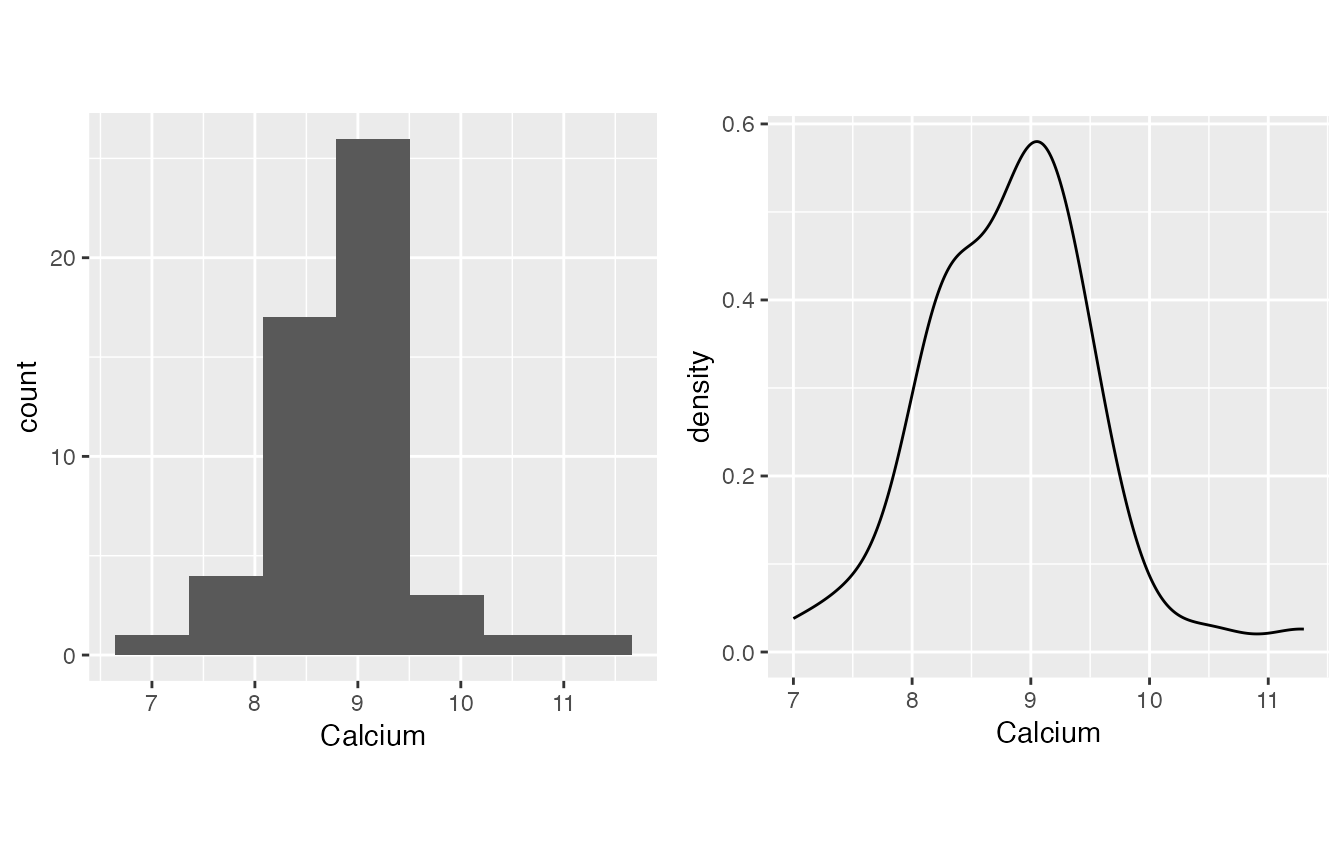
#>
#> $`3`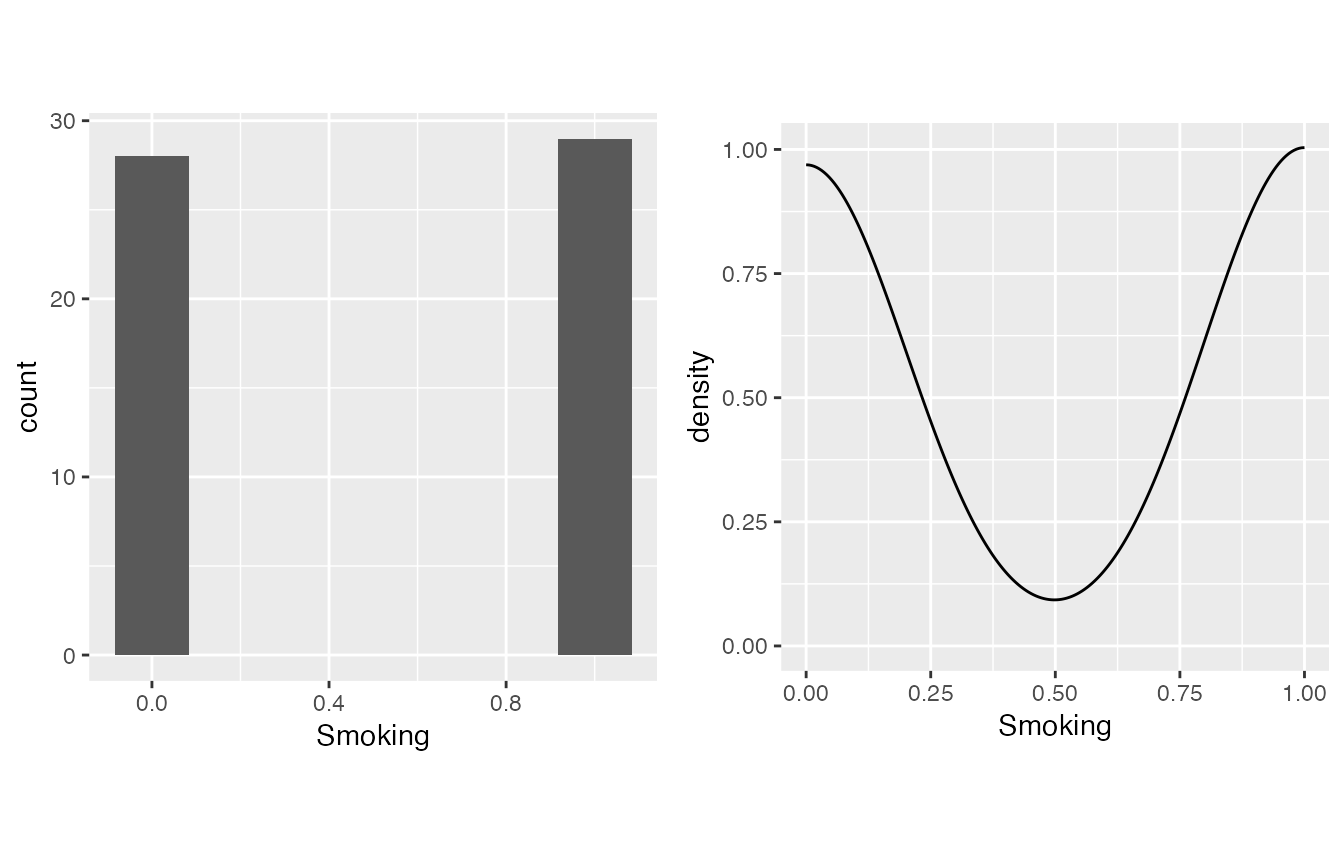
#>
#> $`4`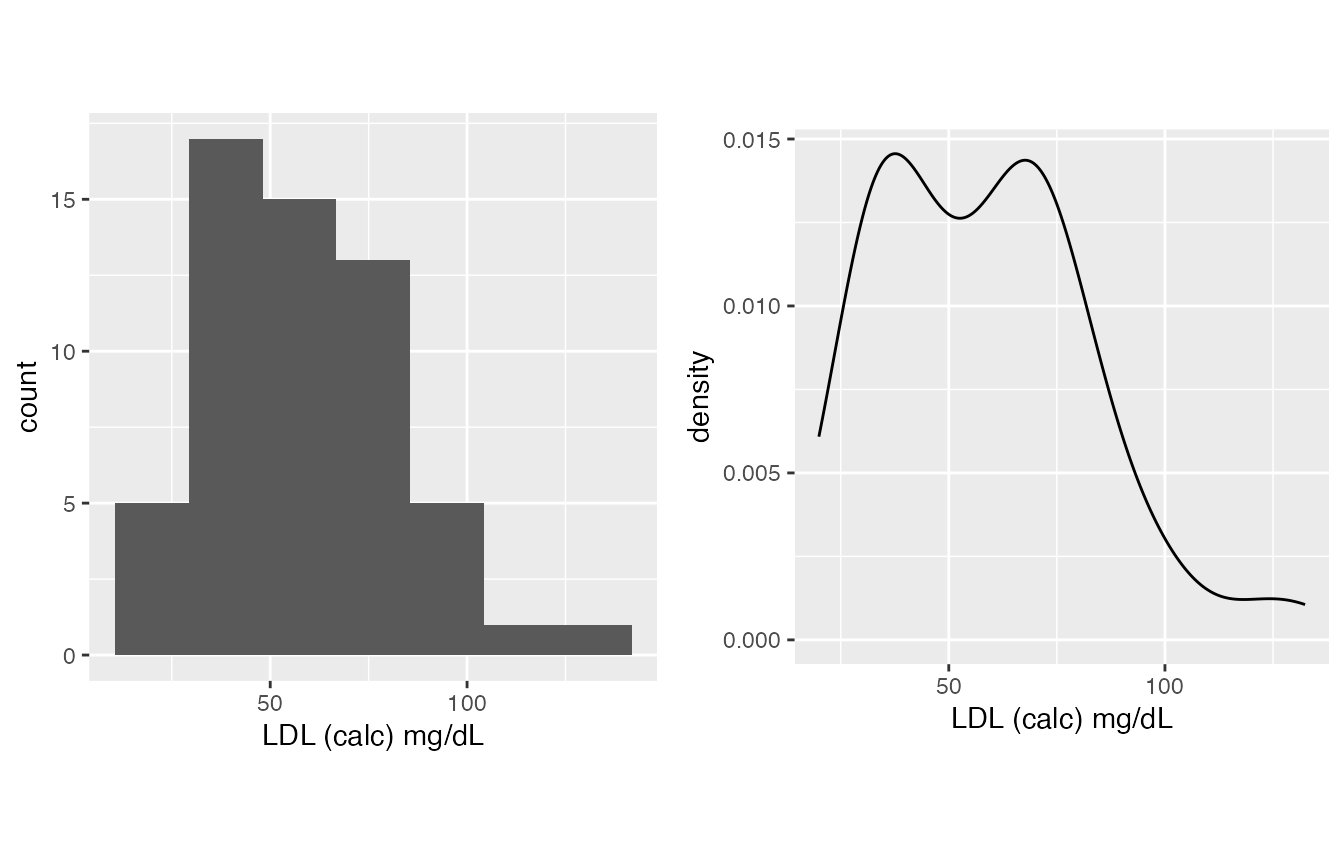
#>
#> $`5`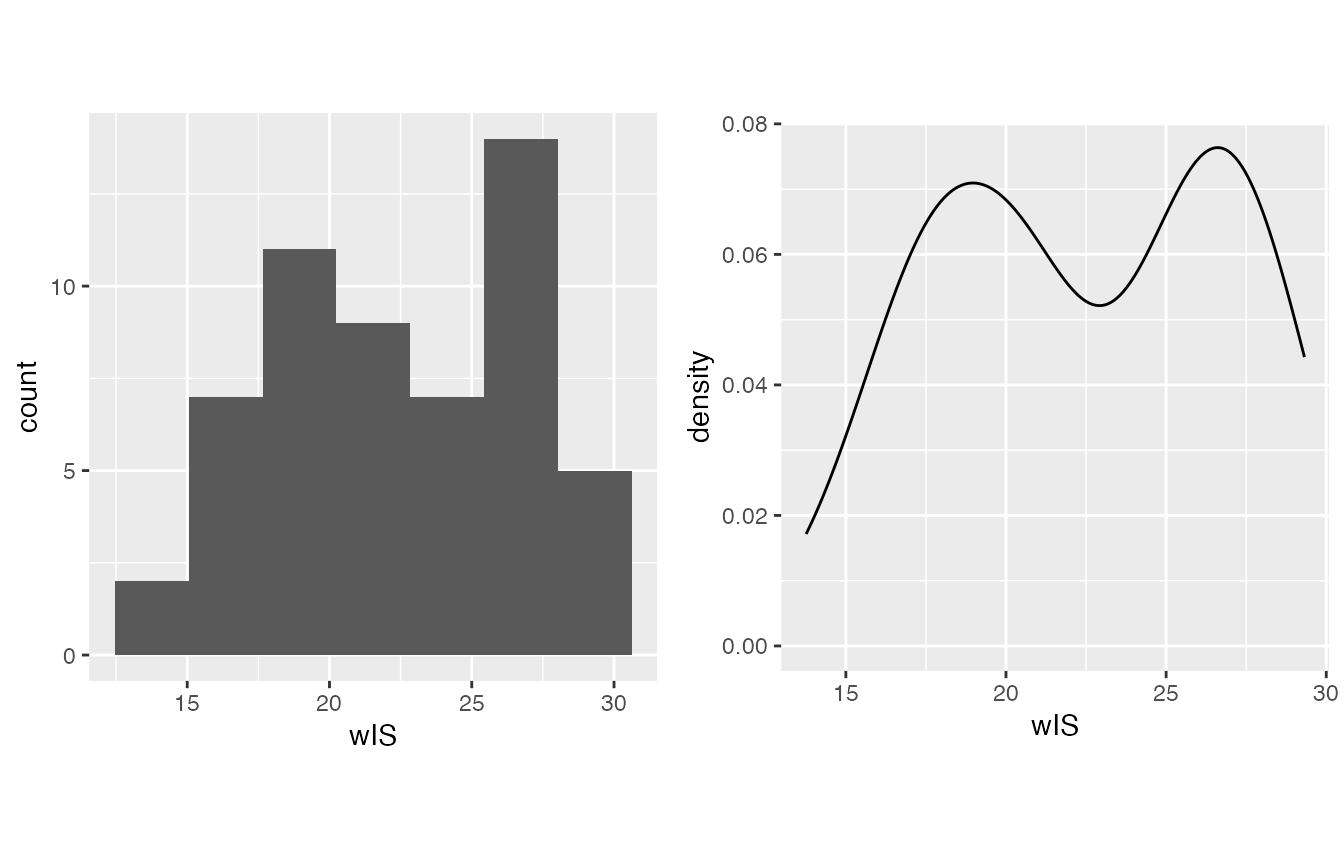
#>
#> $`6`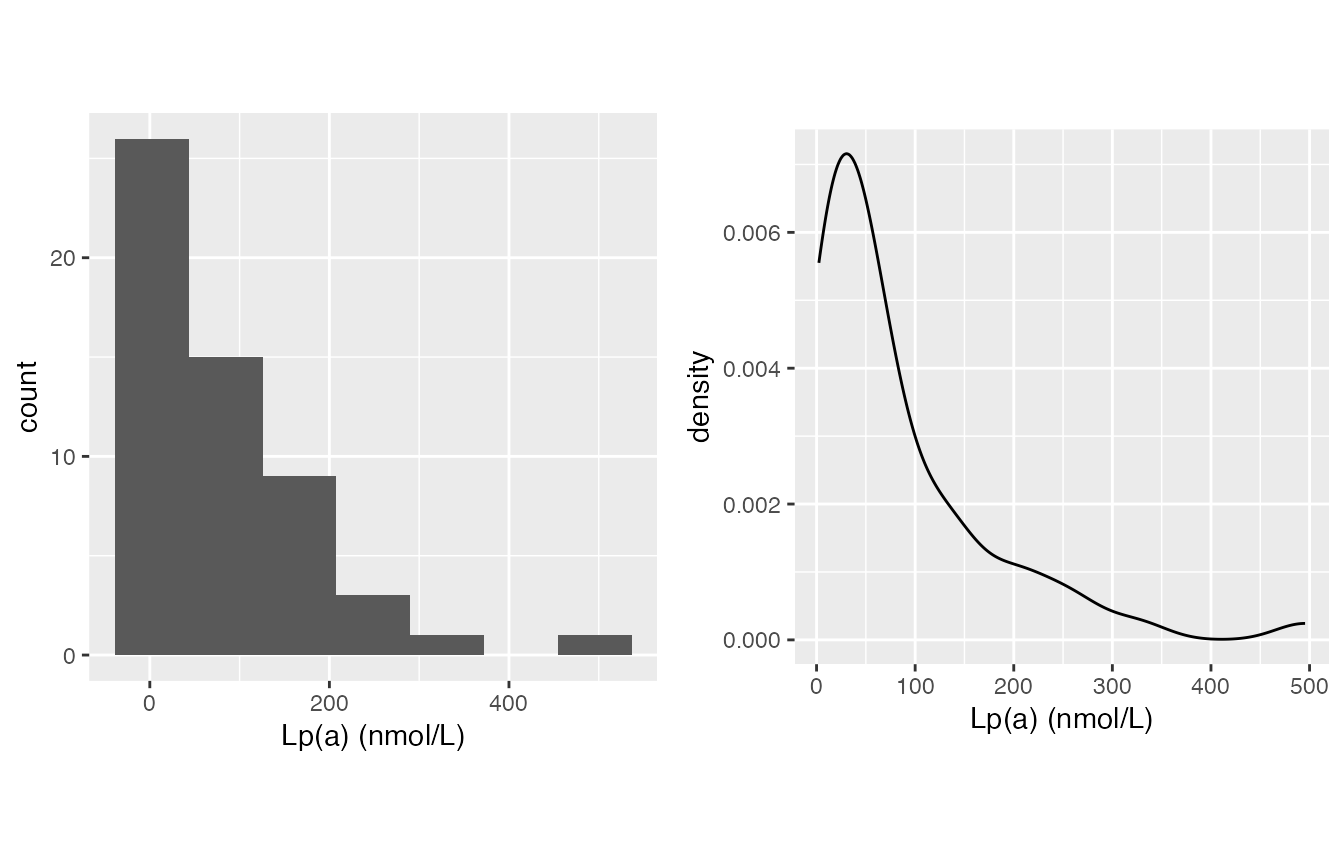
#>
#> attr(,"class")
#> [1] "list" "ggarrange"
#>
#> $`shapiro-wilk_test`
#> variable p_value normality
#> 1 Albumin 2.016173e-03 FALSE
#> 2 Calcium 6.699764e-02 TRUE
#> 3 LDL (calc) mg/dL 1.457113e-02 FALSE
#> 4 Lp(a) (nmol/L) 5.255467e-08 FALSE
#> 5 Smoking 1.278103e-10 FALSE
#> 6 wIS 7.879851e-03 FALSECharacteristic Table
With the result above, you can make your decision and generate a characteristic table.
- Boolean (1/0) data should be in boolean_list, the ratio of 1 will be computed
- Normally distributed continuous data should be in sd_list, the mean(sd) will be computed
- Non-normally distributed continuous data should be in IQR_list, the median(IQR) will be computed
These three lists can be skipped if you don’t have one variable falls into this category.
mirrorstoolkit::characteristic_table_generator(
data = df,
boolean_list = c("Smoking"),
sd_list = c(
"Calcium",
"wIS",
"LDL (calc) mg/dL",
"Albumin"
),
IQR_list = "Lp(a) (nmol/L)",
order = c(
"Smoking",
"Albumin",
"Calcium",
"LDL (calc) mg/dL",
"wIS",
"Lp(a) (nmol/L)")
)
#> overall
#> Smoking 29/57(50.88%)
#> Albumin 3.4 ± 0.63
#> Calcium 8.83 ± 0.73
#> LDL (calc) mg/dL 58.34 ± 24.31
#> wIS 22.46 ± 4.45
#> Lp(a) (nmol/L) 46.76(19.66-118.88)Another implementation can be used here (although I prefer the one above), I don’t know why R original output is not a table for multi-column summary.
Please be aware that this one will run summary() for all columns provided in this table.
mirrorstoolkit::multi_line_summary(
df[,c(
"Smoking",
"Albumin",
"Calcium",
"LDL (calc) mg/dL",
"wIS",
"Lp(a) (nmol/L)")])
#> Warning in (function (..., deparse.level = 1) : number of rows of result is not
#> a multiple of vector length (arg 1)
#> Min. 1st Qu. Median Mean 3rd Qu. Max.
#> Smoking 0.00000 0.00000 1.00000 0.5087719 1.0000 1.0000
#> Albumin 2.10000 3.00000 3.60000 3.3981132 3.9000 4.3000
#> Calcium 7.00000 8.30000 8.90000 8.8339623 9.3000 11.3000
#> LDL (calc) mg/dL 19.95264 36.36183 57.88097 58.3391133 72.3185 132.4144
#> wIS 13.76000 18.37000 21.80000 22.4583636 26.3950 29.3200
#> Lp(a) (nmol/L) 2.36000 19.65500 46.76000 82.5434545 118.8850 495.4500
#> NA's
#> Smoking 0.00000
#> Albumin 4.00000
#> Calcium 4.00000
#> LDL (calc) mg/dL 19.95264
#> wIS 2.00000
#> Lp(a) (nmol/L) 2.00000For two groups result
Most of our analysis is based on two groups, e.g. African American vs Caucasians, Study vs control, etc.
Normality Inspection
Add a “by” variable will classify the data. However, I’d suggest keep watching it overall for the distribution. This is simply for completeness in quick inspection.
mirrorstoolkit::inspect_distribution(
data = df,
by = "Race",
variable = c(
"Albumin",
"Calcium",
"Smoking",
"LDL (calc) mg/dL",
"wIS",
"Lp(a) (nmol/L)")
) %>% base::suppressWarnings()
#> $visualization
#> $`1`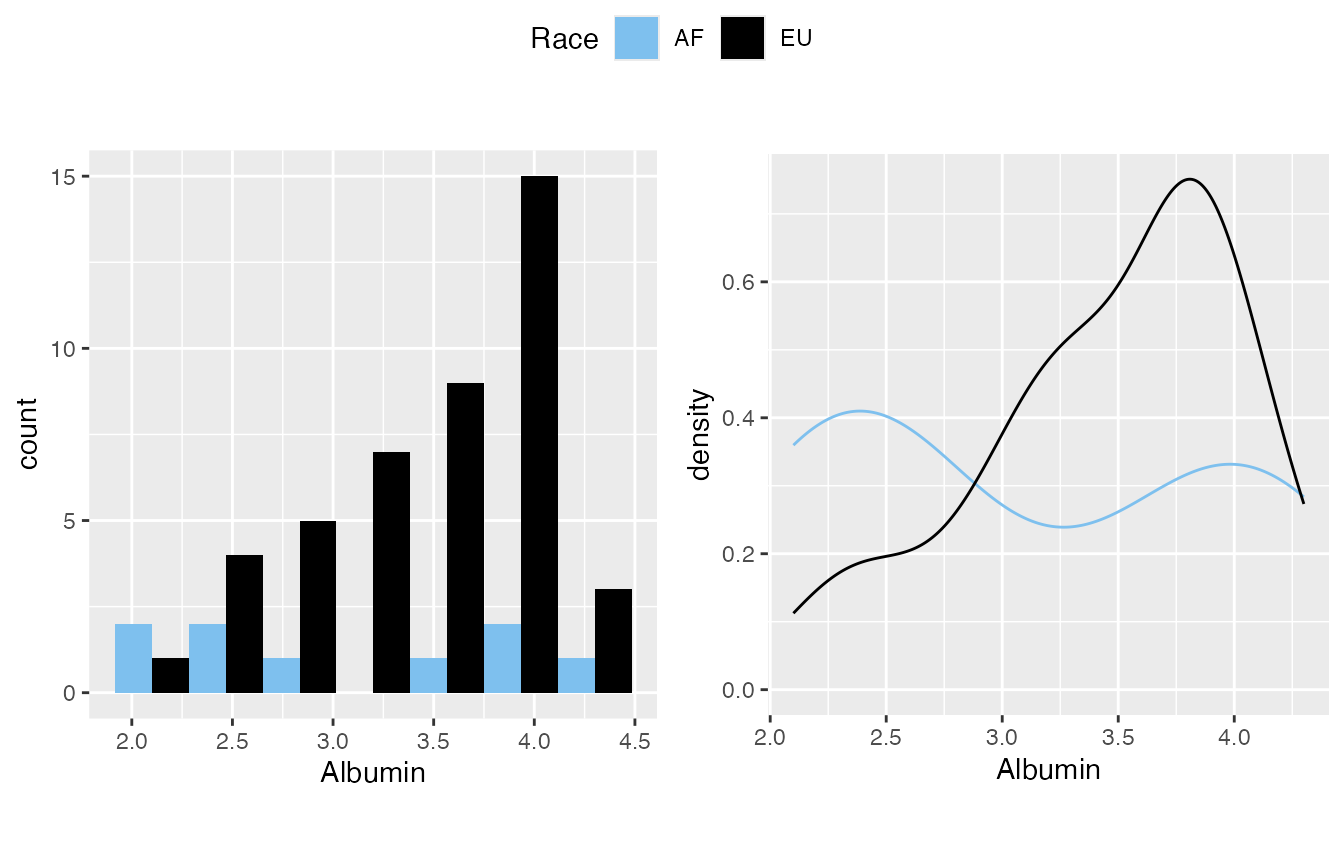
#>
#> $`2`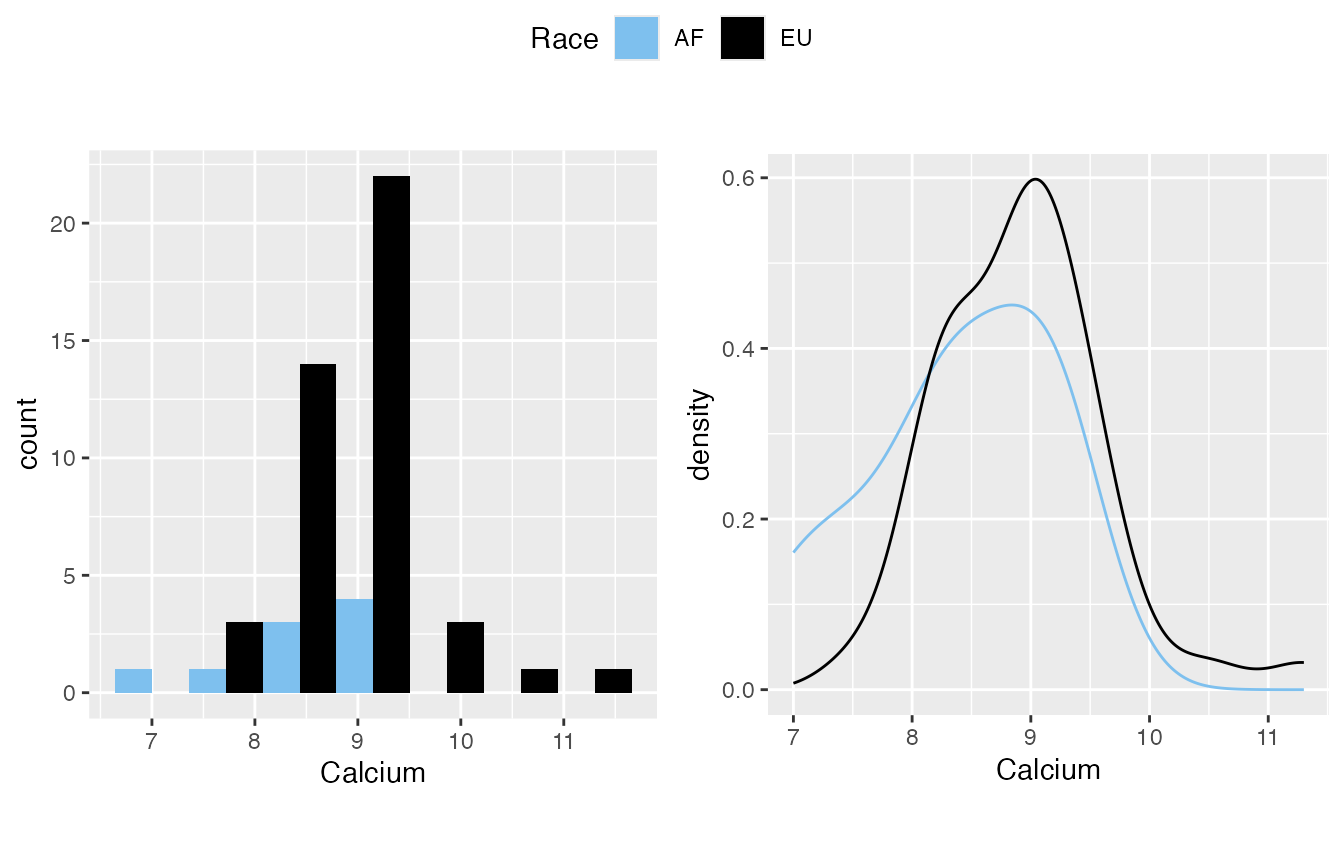
#>
#> $`3`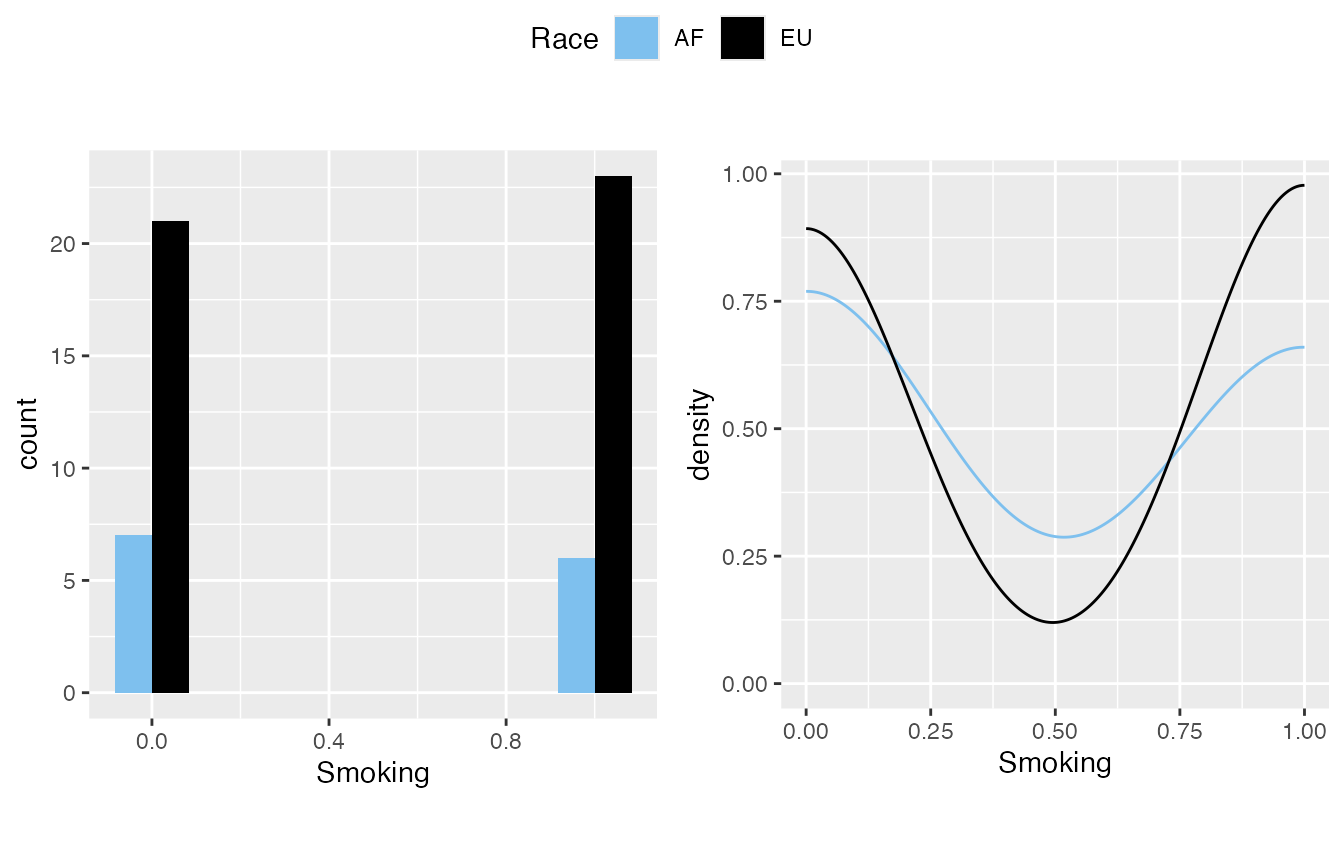
#>
#> $`4`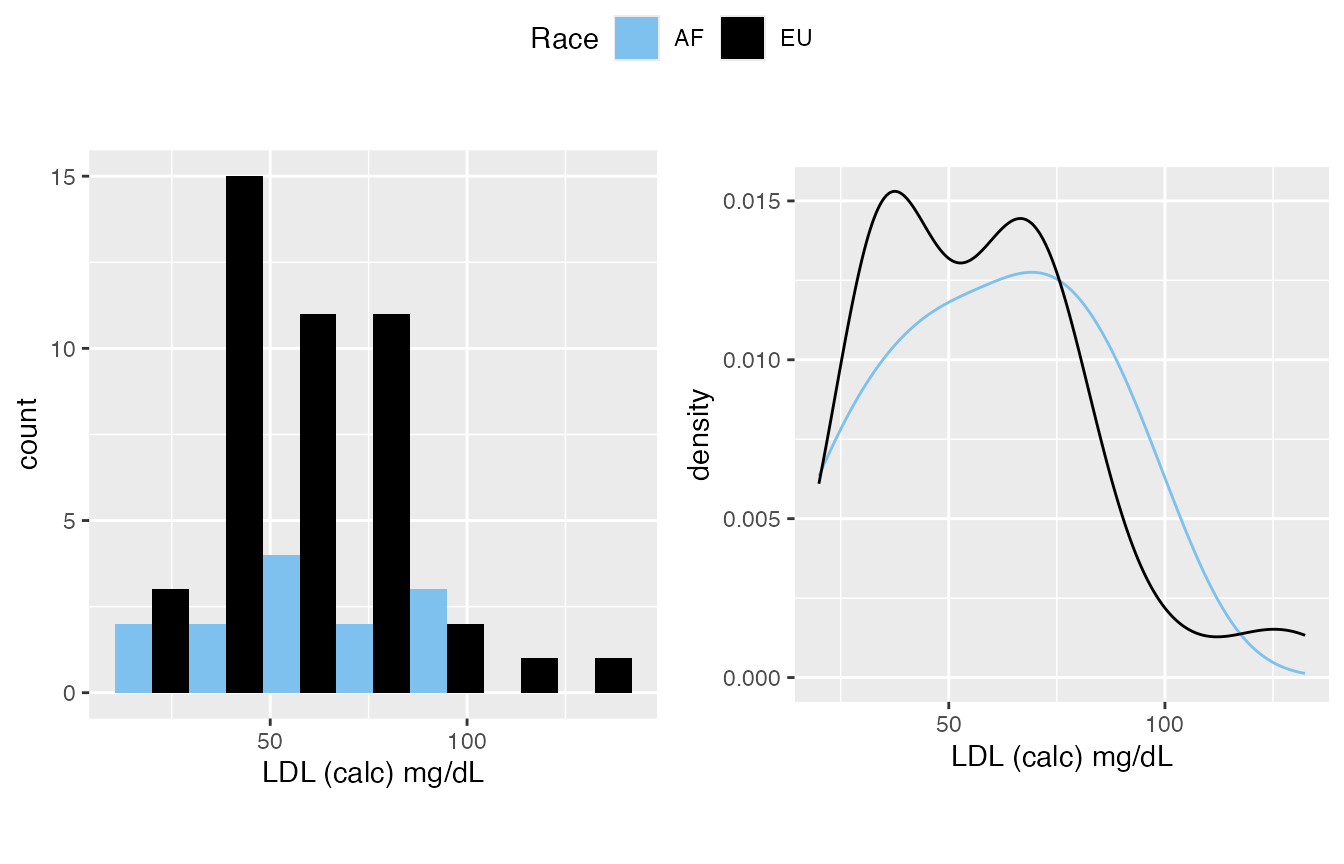
#>
#> $`5`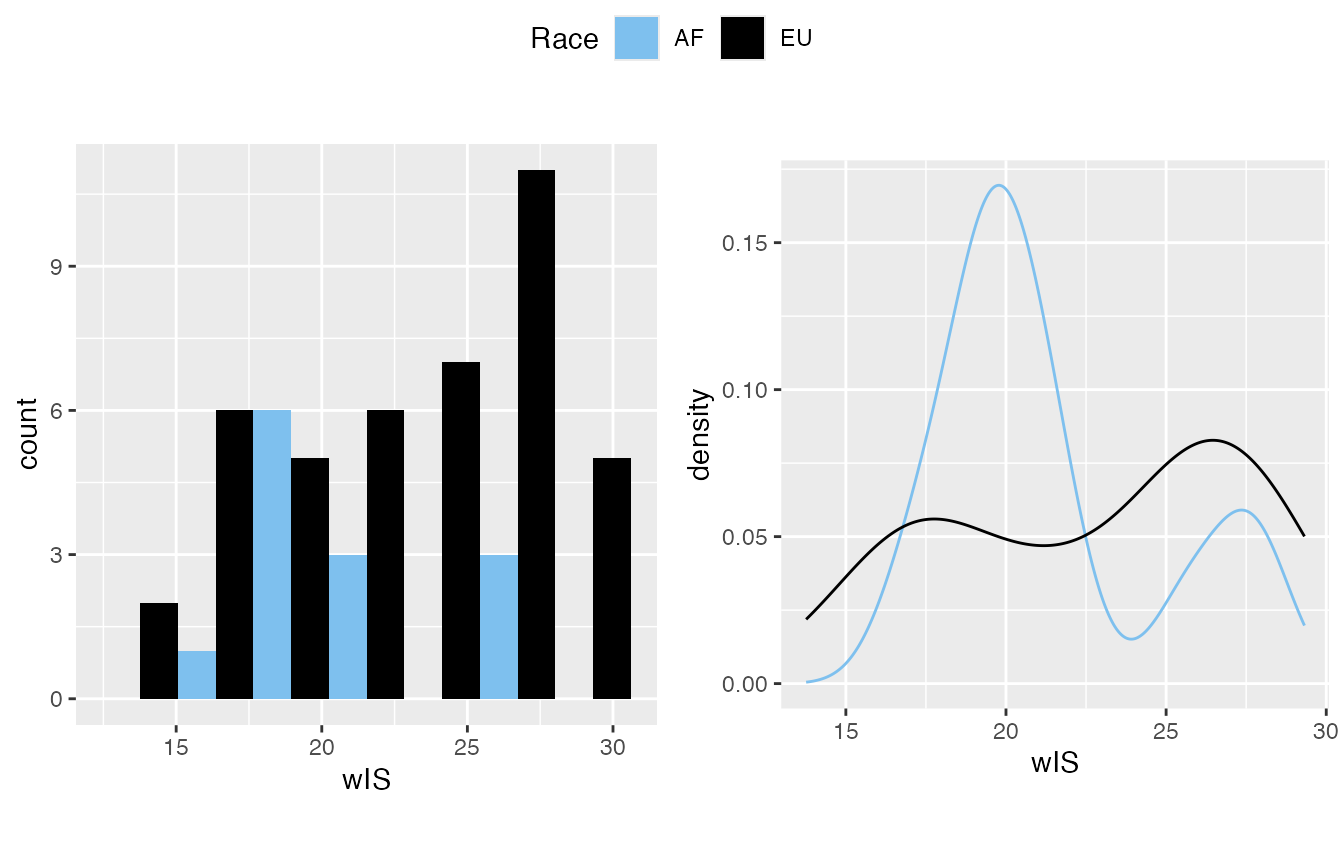
#>
#> $`6`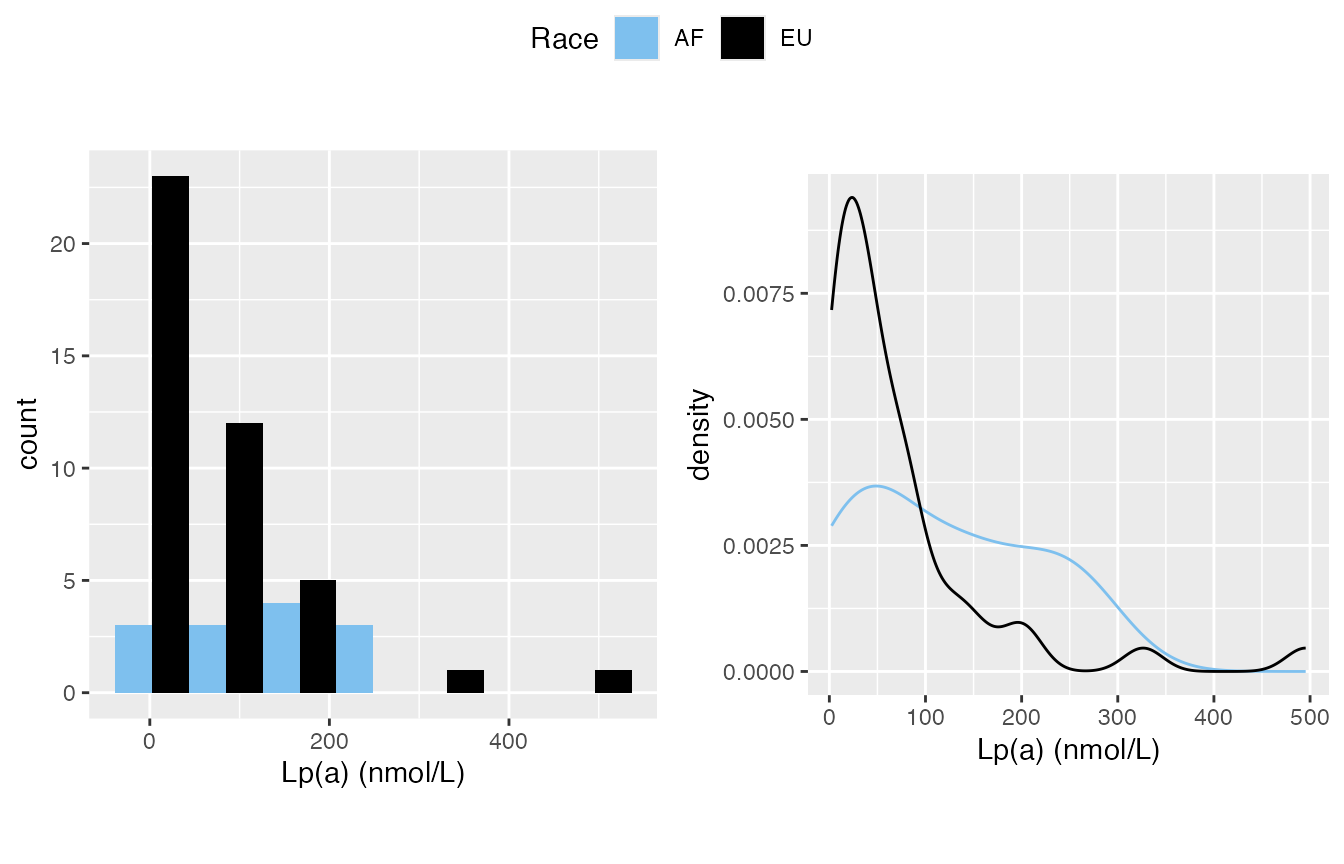
#>
#> attr(,"class")
#> [1] "list" "ggarrange"
#>
#> $`shapiro-wilk_test`
#> variable p_value normality AF EU
#> 1 Albumin 2.016173e-03 FALSE FALSE(3.3e-02) FALSE(1.9e-02)
#> 2 Calcium 6.699764e-02 TRUE TRUE(4.9e-01) FALSE(3.0e-02)
#> 3 LDL (calc) mg/dL 1.457113e-02 FALSE TRUE(9.2e-01) FALSE(3.6e-03)
#> 4 Lp(a) (nmol/L) 5.255467e-08 FALSE TRUE(1.9e-01) FALSE(1.7e-08)
#> 5 Smoking 1.278103e-10 FALSE FALSE(1.7e-04) FALSE(3.4e-09)
#> 6 wIS 7.879851e-03 FALSE FALSE(4.5e-02) FALSE(6.8e-03)Characteristic Table
With a “by” provided, it will generate statistics for sub-class, meanwhile a p_value column is provided.
- For variables in boolean_list, the p-value is from chi-square test (stats::chisq.test())
- For variables in IQR_list, the p-value is from Mann-Whitney U test (Wilcoxon rank-sum test, stats::wilcox.test())
- For varaibles in sd_list, the p-value is depending on variable
normal_policy
- if normal_policy is “wilcox”(default): it will run
Mann-Whitney U test, for the following two reason:
- Our normal data is often not “that normal”.
- A sample size of 30 or more is often cited as a general guideline to ensure a normal distribution is a reasonable approximation.
- if normal_policy is “t-test”: it will run Welch’s t-test (stats::t.test(var.equal = FALSE)), which is a more reliable adaptation of Student’s t-test when the two samples have unequal variances and possibly unequal sample sizes
- if normal_policy is “wilcox”(default): it will run
Mann-Whitney U test, for the following two reason:
mirrorstoolkit::characteristic_table_generator(
data = df,
by = "Race",
boolean_list = c("Smoking"),
sd_list = c(
"Calcium",
"wIS",
"LDL (calc) mg/dL",
"Albumin"
),
IQR_list = "Lp(a) (nmol/L)",
order = c(
"Smoking",
"Albumin",
"Calcium",
"LDL (calc) mg/dL",
"wIS",
"Lp(a) (nmol/L)"),
normal_policy = "t-test"
)
#> overall AF EU
#> Smoking 29/57(50.88%) 6/13(46.15%) 23/44(52.27%)
#> Albumin 3.4 ± 0.63 3.1 ± 0.88 3.46 ± 0.56
#> Calcium 8.83 ± 0.73 8.41 ± 0.79 8.92 ± 0.7
#> LDL (calc) mg/dL 58.34 ± 24.31 60.24 ± 24.85 57.78 ± 24.41
#> wIS 22.46 ± 4.45 21.31 ± 3.52 22.81 ± 4.68
#> Lp(a) (nmol/L) 46.76(19.66-118.88) 127.64(46.92-197.37) 41.08(15.27-79.61)
#> p_value
#> Smoking 7.5912e-01
#> Albumin 2.6747e-01
#> Calcium 9.9941e-02
#> LDL (calc) mg/dL 7.5591e-01
#> wIS 2.2530e-01
#> Lp(a) (nmol/L) 4.8697e-02For 3+ groups result
Characteristic Table
With a “by” provided, it will generate statistics for sub-class, meanwhile a p_value column is provided.
- For variables in boolean_list, the p-value is from chi-square test (stats::chisq.test())
- For variables in IQR_list, the p-value is from Kruskal–Wallis test (stats::kruskal.test())
- For varaibles in sd_list, the p-value is depending on variable
normal_policy
- if normal_policy is “wilcox”(default, I inherited the name
from above thus won’t make things too complex): it will run
stats::kruskal.test(), for the following two reason:
- Our normal data is often not “that normal”.
- A sample size of 30 or more is often cited as a general guideline to ensure a normal distribution is a reasonable approximation.
- if normal_policy is “t-test”: it will run ANOVA(stats::aov())
- if normal_policy is “wilcox”(default, I inherited the name
from above thus won’t make things too complex): it will run
stats::kruskal.test(), for the following two reason:
df = df %>% dplyr::mutate(
Calciphylaxis_str = Calciphylaxis %>% plyr::mapvalues(c(0,1), c("control", "disease")),
group = paste(`Race`, `Calciphylaxis_str`, sep = "_"))
df$group %>% table()
#> .
#> AF_control AF_disease EU_control EU_disease
#> 8 5 22 22
table_1_by_group = mirrorstoolkit::characteristic_table_generator(
data = df,
by = "group",
boolean_list = c("Smoking"),
sd_list = c(
"Calcium",
"wIS",
"LDL (calc) mg/dL",
"Albumin"
),
IQR_list = "Lp(a) (nmol/L)",
order = c(
"Smoking",
"Albumin",
"Calcium",
"LDL (calc) mg/dL",
"wIS",
"Lp(a) (nmol/L)"),
normal_policy = "t-test"
)
table_1_by_group
#> overall AF_control AF_disease
#> Smoking 29/57(50.88%) 4/8(50%) 2/5(40%)
#> Albumin 3.4 ± 0.63 3.52 ± 0.83 2.76 ± 0.83
#> Calcium 8.83 ± 0.73 8.7 ± 0.45 8.18 ± 0.97
#> LDL (calc) mg/dL 58.34 ± 24.31 64.04 ± 28.44 54.16 ± 19.04
#> wIS 22.46 ± 4.45 22.22 ± 3.48 19.85 ± 3.4
#> Lp(a) (nmol/L) 46.76(19.66-118.88) 137.46(43.7-206.76) 55.59(51.67-129.45)
#> EU_control EU_disease p_value
#> Smoking 12/22(54.55%) 11/22(50%) 9.8051e-01
#> Albumin 3.82 ± 0.3 3.1 ± 0.54 2.8352e-05
#> Calcium 8.79 ± 0.54 9.05 ± 0.82 1.0060e-01
#> LDL (calc) mg/dL 56.18 ± 17.64 59.38 ± 30.07 8.5607e-01
#> wIS 22.98 ± 4.51 22.65 ± 4.96 5.7002e-01
#> Lp(a) (nmol/L) 33.85(18.76-87.43) 43.4(14.11-75.14) 2.0291e-01The output illutrated the p-value as literal string for I need to keep the format for paper use… To filter the p-value you can do this
Pairwise test
Please always be aware that for 3+ group test, one p-value is not sufficient to claim too much stuff. Multi-class chi-square/Kruskal-Wallis/ANOVA only told you “some group is different from the other”
Boolean Variables
Pairwise comparisons between pairs of proportions (stats::pairwise.prop.test) used here, a filled barchart used in the visualization.
When the pairwise test is throwing “Warning: Chi-squared approximation may be incorrect” means that one or more of your table’s expected cell counts are too small for the Chi-squared test to be reliable, typically when they are less than 5. This violates the test’s assumptions, and you should address it by either increasing sample size, combining categories, or using an alternative like Fisher’s exact test or a simulated p-value.
- If it’s labelled significant, it can be a good support to your claim.
- If it’s not labelled as significant, it doesn’t always mean that they are not different, it’s simply stats::pairwise.prop.test cannot handle it. Be bold to make your choice.
Some settings, I grabbed a setting from the air for the illustration:
- adjustment_method The choice of this variable is from here. “fdr” (Benjamini-Hochberg FDR correction) by default thus you can often skip it.
- p_threshold will affect the filtering from first to second table, the significant result will be labelled on the top of the figure, 0.05 by default
- variable can take one or more variables.
pairwise_boolean = mirrorstoolkit::pairwise_boolean_report(
data = df,
variable = c("antiplatelet_group", "statin_group"),
by = "group",
adjustment_method = "none",
p_threshold = 0.3)
#> Warning in prop.test(x[c(i, j)], n[c(i, j)], ...): Chi-squared approximation
#> may be incorrect
#> Warning in prop.test(x[c(i, j)], n[c(i, j)], ...): Chi-squared approximation
#> may be incorrect
#> Warning in prop.test(x[c(i, j)], n[c(i, j)], ...): Chi-squared approximation
#> may be incorrect
#> Warning in prop.test(x[c(i, j)], n[c(i, j)], ...): Chi-squared approximation
#> may be incorrect
#> Warning in prop.test(x[c(i, j)], n[c(i, j)], ...): Chi-squared approximation
#> may be incorrect
#> Warning in prop.test(x[c(i, j)], n[c(i, j)], ...): Chi-squared approximation
#> may be incorrect
#> Warning in prop.test(x[c(i, j)], n[c(i, j)], ...): Chi-squared approximation
#> may be incorrect
#> Warning in prop.test(x[c(i, j)], n[c(i, j)], ...): Chi-squared approximation
#> may be incorrect
#> Warning in prop.test(x[c(i, j)], n[c(i, j)], ...): Chi-squared approximation
#> may be incorrect
#> Warning in prop.test(x[c(i, j)], n[c(i, j)], ...): Chi-squared approximation
#> may be incorrect
#> Warning in ggsignif::geom_signif(data = annotation_significant,
#> ggplot2::aes(xmin = group1, : Ignoring unknown aesthetics: xmin,
#> xmax, annotations, and y_position
pairwise_boolean$result
#> # A tibble: 12 × 4
#> variable_name group1 group2 p_value
#> <chr> <chr> <chr> <dbl>
#> 1 antiplatelet_group AF_disease AF_control 0.396
#> 2 antiplatelet_group EU_control AF_control 1.00
#> 3 antiplatelet_group EU_control AF_disease 0.357
#> 4 antiplatelet_group EU_disease AF_control 1
#> 5 antiplatelet_group EU_disease AF_disease 0.281
#> 6 antiplatelet_group EU_disease EU_control 0.931
#> 7 statin_group AF_disease AF_control 1
#> 8 statin_group EU_control AF_control 1
#> 9 statin_group EU_control AF_disease 1
#> 10 statin_group EU_disease AF_control 0.907
#> 11 statin_group EU_disease AF_disease 1
#> 12 statin_group EU_disease EU_control 0.803
pairwise_boolean$significant_result
#> # A tibble: 1 × 4
#> variable_name group1 group2 p_value
#> <chr> <chr> <chr> <dbl>
#> 1 antiplatelet_group EU_disease AF_disease 0.28
pairwise_boolean$plot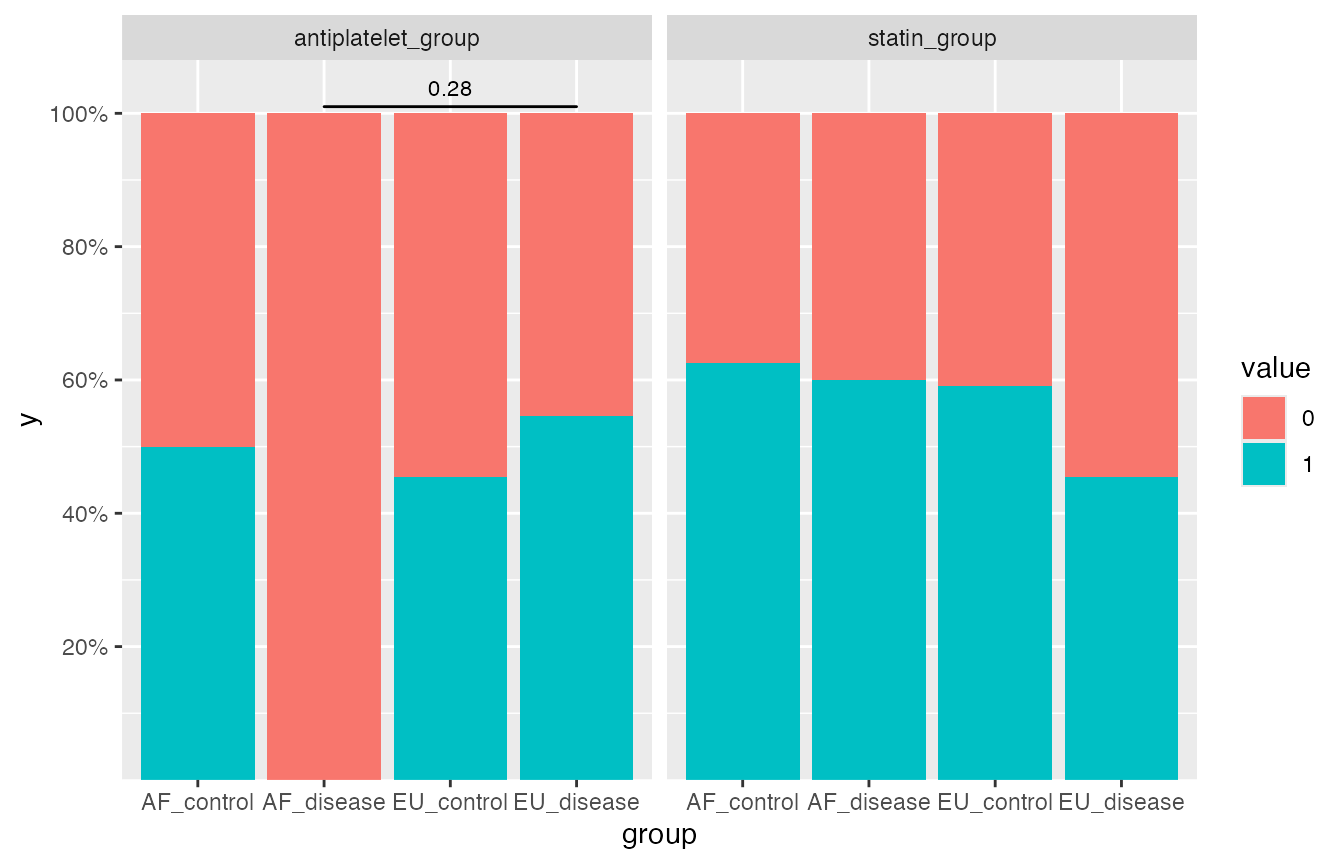
Not Normally Distributed Continuous Variables
Pairwise Mann-Whiteney U-test (stats::pairwise.wilcox.test) used here, boxplot for illustration.
If you are not confident with the normality or the sample size, use this one.
Please be aware of a toxic chemical reaction here:
- Mann-Whiteney U test is based on the rank of the sample
- Boxplot can drop some outliers out.
Thus this report is very visually misleading with extremely small sample size, e.g. AF_disease group only have 5 samples and 1 outliers makes it not significantly different from the other
df$`Lp(a) (nmol/L)`
#> [1] 204.60 7.77 30.60 NA 18.76 86.02 33.85 6.33 31.30 33.85
#> [11] 38.89 58.27 9.88 29.37 252.60 34.02 10.87 46.92 147.29 127.64
#> [21] 234.93 13.59 197.37 87.43 495.45 3.97 125.88 156.62 74.34 195.71
#> [31] 20.55 78.89 43.26 75.14 129.45 3.37 55.59 4.65 8.23 79.85
#> [41] 111.89 43.40 46.76 51.67 33.39 4.65 20.83 44.61 14.11 55.33
#> [51] 326.37 74.65 2.36 273.65 NA 139.47 3.65
pairwise_wilcox = mirrorstoolkit::pairwise_wilcox_report(
data = df,
variable = "Lp(a) (nmol/L)",
by = "group",
p_threshold = 0.30
)
#> Warning in ggsignif::geom_signif(data = annotation_significant,
#> ggplot2::aes(xmin = group1, : Ignoring unknown aesthetics: xmin,
#> xmax, annotations, and y_position
pairwise_wilcox$result
#> # A tibble: 6 × 4
#> variable_name group1 group2 p_value
#> <chr> <chr> <chr> <dbl>
#> 1 Lp(a) (nmol/L) AF_disease AF_control 0.826
#> 2 Lp(a) (nmol/L) EU_control AF_control 0.250
#> 3 Lp(a) (nmol/L) EU_control AF_disease 0.738
#> 4 Lp(a) (nmol/L) EU_disease AF_control 0.250
#> 5 Lp(a) (nmol/L) EU_disease AF_disease 0.738
#> 6 Lp(a) (nmol/L) EU_disease EU_control 0.738
pairwise_wilcox$significant_result
#> # A tibble: 2 × 4
#> variable_name group1 group2 p_value
#> <chr> <chr> <chr> <dbl>
#> 1 Lp(a) (nmol/L) EU_control AF_control 0.25
#> 2 Lp(a) (nmol/L) EU_disease AF_control 0.25
pairwise_wilcox$plot
#> Warning: Removed 2 rows containing non-finite outside the scale range
#> (`stat_boxplot()`).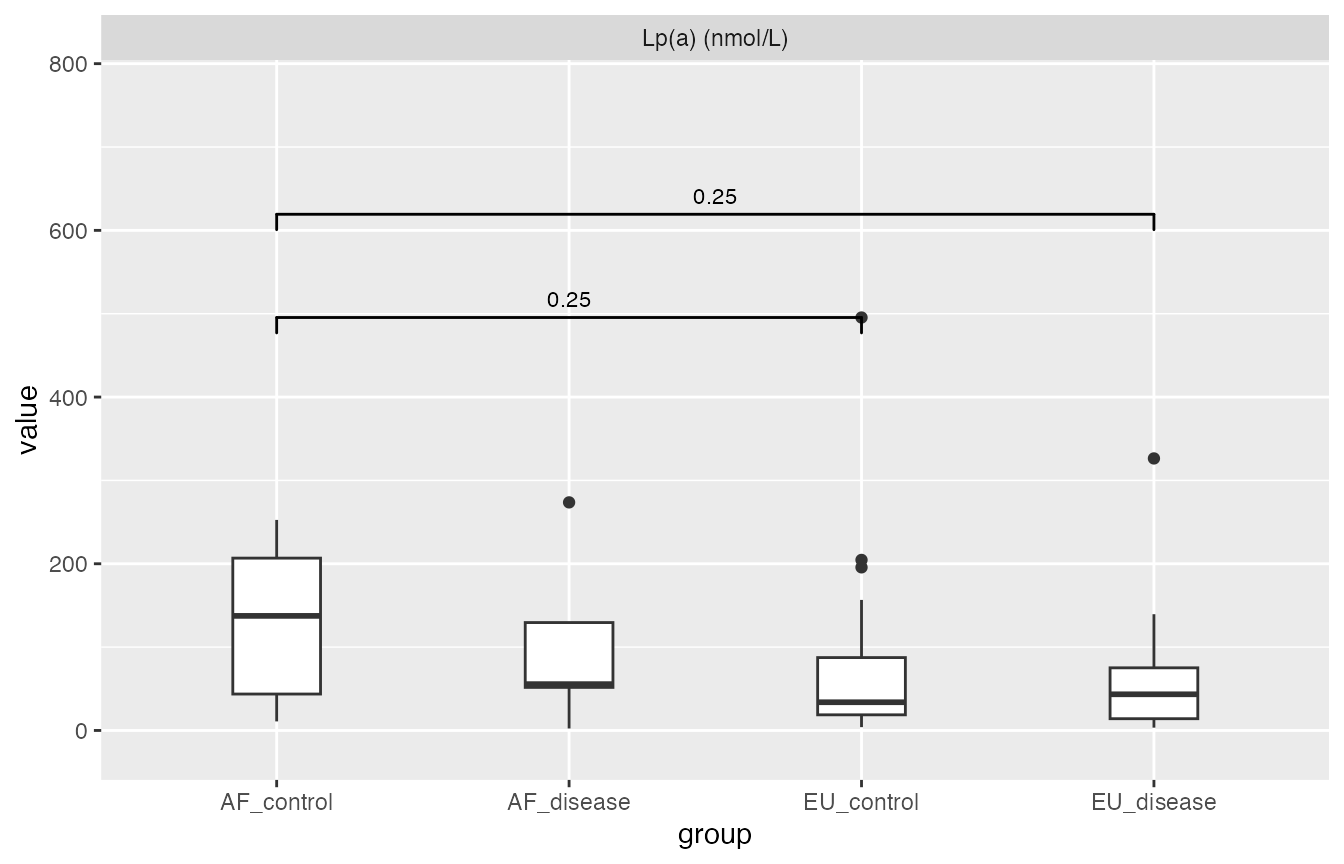
Normally Distributed Continuous Variables
Pairwise t-test (stats::pairwise.t.test) used here, barchart for mean with errorbar (1 sd, not 95% CI) for illustration.
Still, if you are not confident with the normality or the sample size, use the one for not normally distributed variable above.
pairwise_t_test = mirrorstoolkit::pairwise_t_test_report(
data = df,
variable = c("Calcium","Albumin"),
by = "group",
p_threshold = 0.15
)
#> Warning in ggsignif::geom_signif(data = annotation_significant,
#> ggplot2::aes(xmin = group1, : Ignoring unknown aesthetics: xmin,
#> xmax, annotations, and y_position
pairwise_t_test$result
#> # A tibble: 12 × 4
#> variable_name group1 group2 p_value
#> <chr> <chr> <chr> <dbl>
#> 1 Albumin AF_disease AF_control 0.0617
#> 2 Albumin EU_control AF_control 0.298
#> 3 Albumin EU_control AF_disease 0.000379
#> 4 Albumin EU_disease AF_control 0.201
#> 5 Albumin EU_disease AF_disease 0.225
#> 6 Albumin EU_disease EU_control 0.000156
#> 7 Calcium AF_disease AF_control 0.416
#> 8 Calcium EU_control AF_control 0.814
#> 9 Calcium EU_control AF_disease 0.261
#> 10 Calcium EU_disease AF_control 0.439
#> 11 Calcium EU_disease AF_disease 0.0977
#> 12 Calcium EU_disease EU_control 0.416
pairwise_t_test$significant_result
#> # A tibble: 4 × 4
#> variable_name group1 group2 p_value
#> <chr> <chr> <chr> <dbl>
#> 1 Albumin AF_disease AF_control 0.062
#> 2 Albumin EU_control AF_disease 0.00038
#> 3 Albumin EU_disease EU_control 0.00016
#> 4 Calcium EU_disease AF_disease 0.098
pairwise_t_test$plot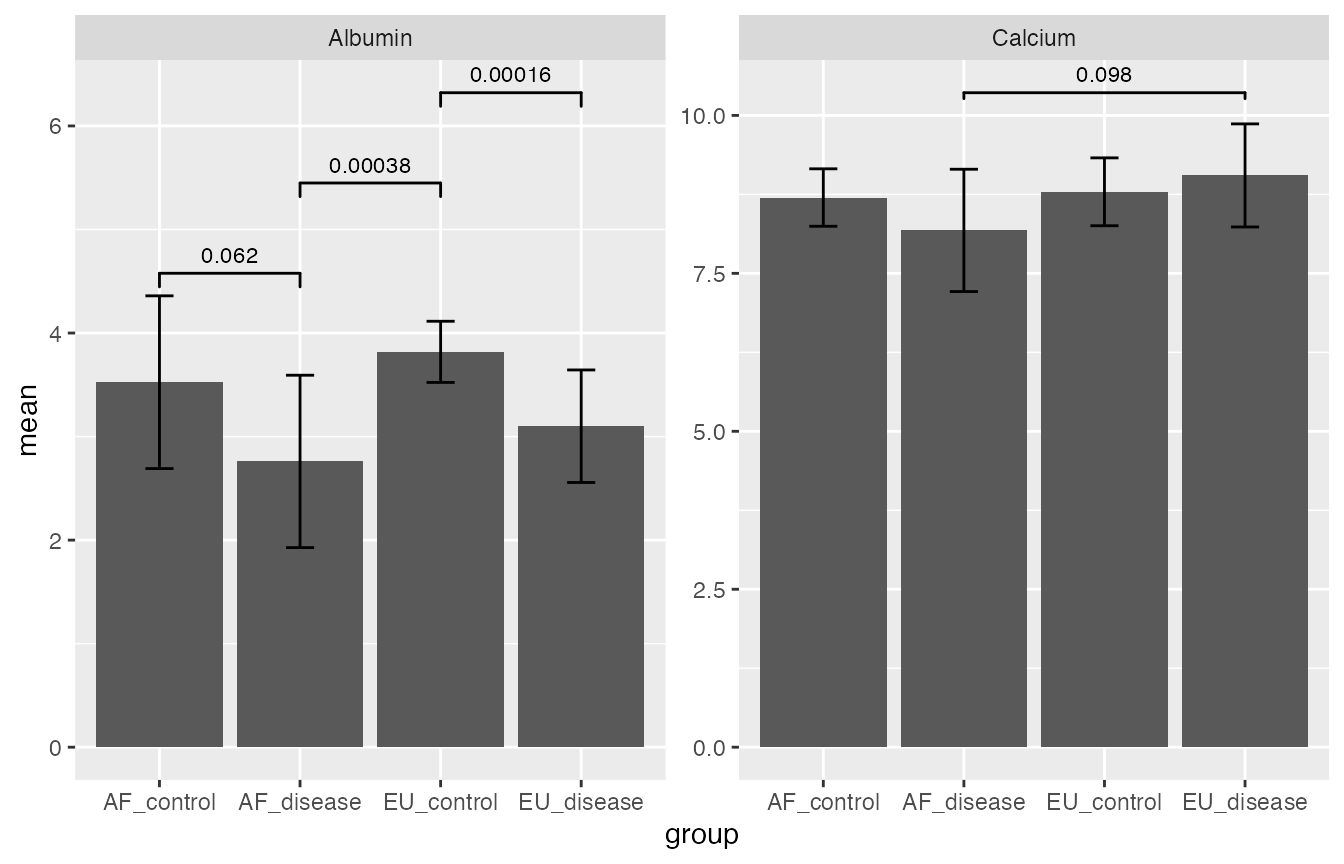
If you only want the first table in supplemental data, you can run the one below as well. Replace “report” to “table” in the function name and remove the p_threshold if you have. This function is internally called in mirrorstoolkit::pairwise_t_test_report, so do the other two.
mirrorstoolkit::pairwise_t_test_table(
data = df,
variable = "Calcium",
by = "group"
)
#> # A tibble: 6 × 4
#> variable_name group1 group2 p_value
#> <chr> <chr> <chr> <dbl>
#> 1 Calcium AF_disease AF_control 0.416
#> 2 Calcium EU_control AF_control 0.814
#> 3 Calcium EU_control AF_disease 0.261
#> 4 Calcium EU_disease AF_control 0.439
#> 5 Calcium EU_disease AF_disease 0.0977
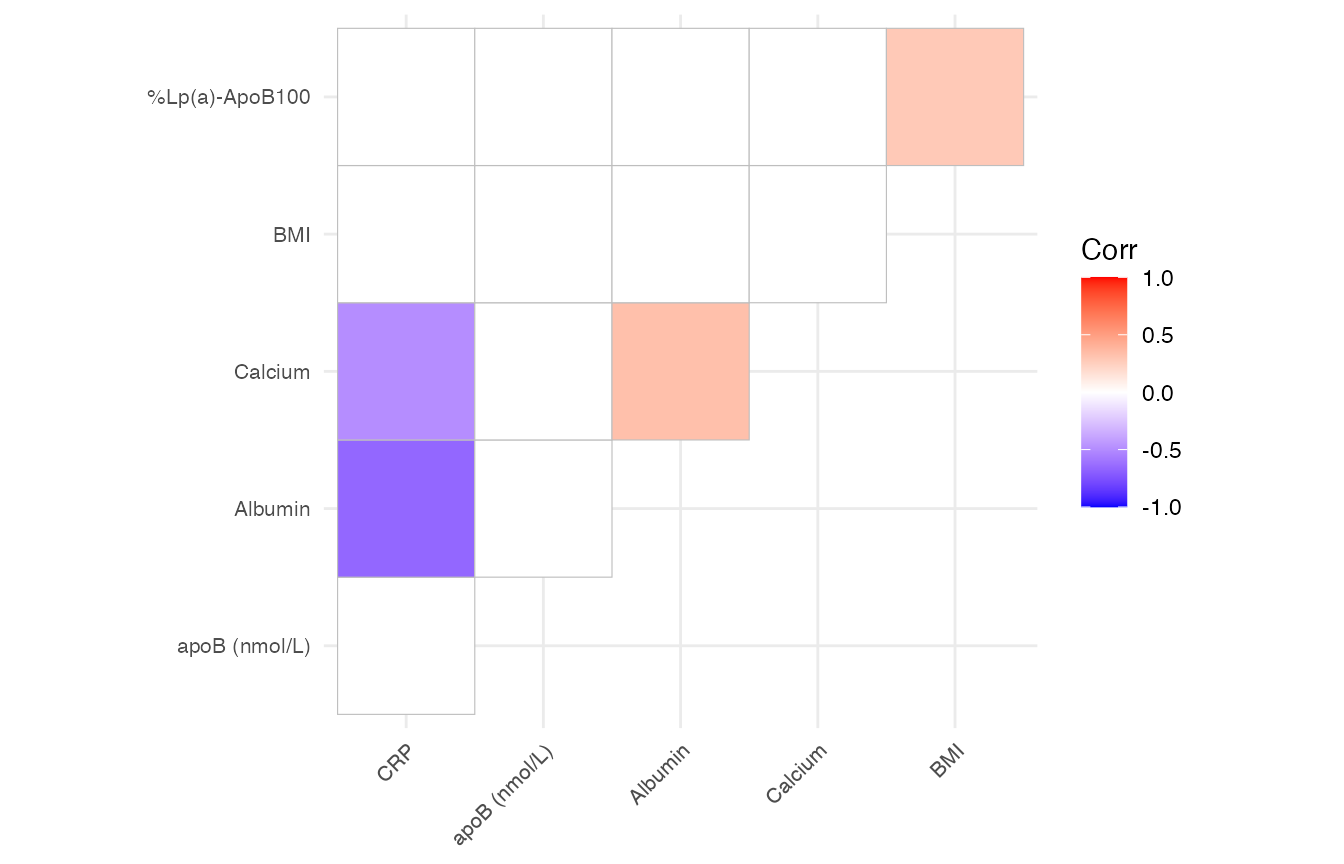
#> 6 Calcium EU_disease EU_control 0.416Correlation matrix
We provide two functions for correlation matrix analysis.
To visualize the correlation matrix
mirrorstoolkit::correlation_matrix_plot will return a ggplot2 instance, sharing a similar argument setting:
- variable is optional, if not provided, will use all the columns provided in data. However, as a fool-proofing operation, providing the variable column is a good sanity check as the function will internally enforces all the value in your data table to numeric.
- font_size is 5 by default for all labels.
- please be aware that the order is reordered by hierarchical clustering for better cluster illustration
mirrorstoolkit::correlation_matrix_plot(
data = df,
variable = c("BMI", "Albumin", "Calcium", "CRP", "apoB (nmol/L)","%Lp(a)-ApoB100"),
font_size = 8
)
#> Warning: `aes_string()` was deprecated in ggplot2 3.0.0.
#> ℹ Please use tidy evaluation idioms with `aes()`.
#> ℹ See also `vignette("ggplot2-in-packages")` for more information.
#> ℹ The deprecated feature was likely used in the ggcorrplot package.
#> Please report the issue at <https://github.com/kassambara/ggcorrplot/issues>.
#> This warning is displayed once per session.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
#> generated.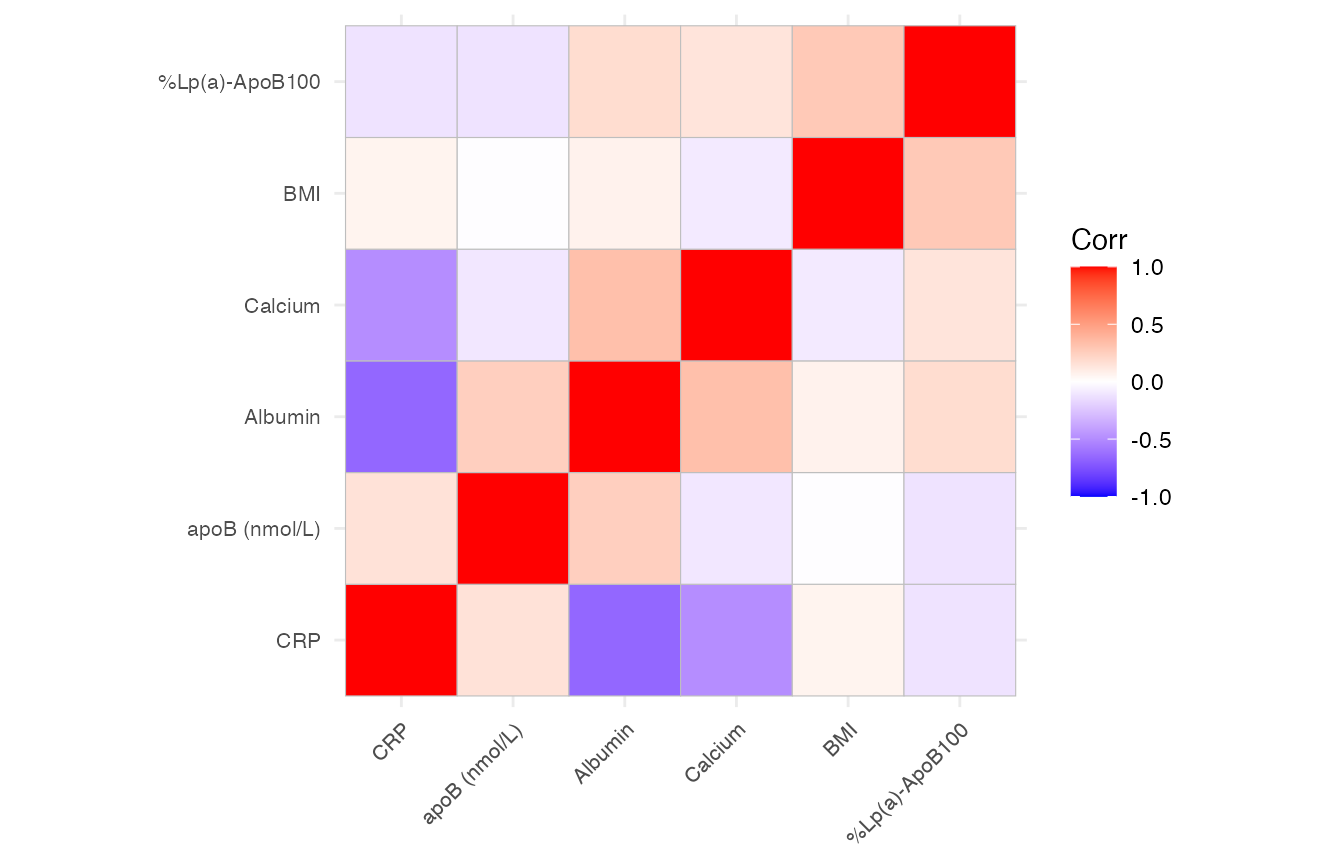
This function also provide a filter operation by the p-value as long as the p_threshold is specified. see below (I changed the way inputting column here for illustration purpose, it doesn’t matter):
mirrorstoolkit::correlation_matrix_plot(
data = df %>% dplyr::select(
tidyr::all_of(c("BMI", "Albumin", "Calcium", "CRP", "apoB (nmol/L)","%Lp(a)-ApoB100"))
),
font_size = 8,
p_threshold = 0.05
)
To further deal with the correlation matrix
For potential multi-omics studies in the future, I left an function here outputting the matrix as data.frame format, which removed all the redundant columns. It has a p_threshold option same as above but I don’t think it’s necessary here.
mirrorstoolkit::correlation_matrix_dataframe(
data = df,
variable = c("BMI", "Albumin", "Calcium", "CRP", "apoB (nmol/L)","%Lp(a)-ApoB100")
)
#> row column corr p_value
#> 1 BMI Albumin 0.07246750 6.096735e-01
#> 2 BMI Calcium -0.09116465 5.203815e-01
#> 3 Albumin Calcium 0.33290055 1.486389e-02
#> 4 BMI CRP 0.06434398 7.309315e-01
#> 5 Albumin CRP -0.66165518 3.729085e-05
#> 6 Calcium CRP -0.48625030 4.776512e-03
#> 7 BMI apoB (nmol/L) -0.00519316 9.696990e-01
#> 8 Albumin apoB (nmol/L) 0.25441832 6.600239e-02
#> 9 Calcium apoB (nmol/L) -0.10436060 4.570736e-01
#> 10 CRP apoB (nmol/L) 0.15435436 3.989530e-01
#> 11 BMI %Lp(a)-ApoB100 0.27960245 4.059818e-02
#> 12 Albumin %Lp(a)-ApoB100 0.17946329 2.076293e-01
#> 13 Calcium %Lp(a)-ApoB100 0.14327436 3.158588e-01
#> 14 CRP %Lp(a)-ApoB100 -0.11916085 5.231636e-01
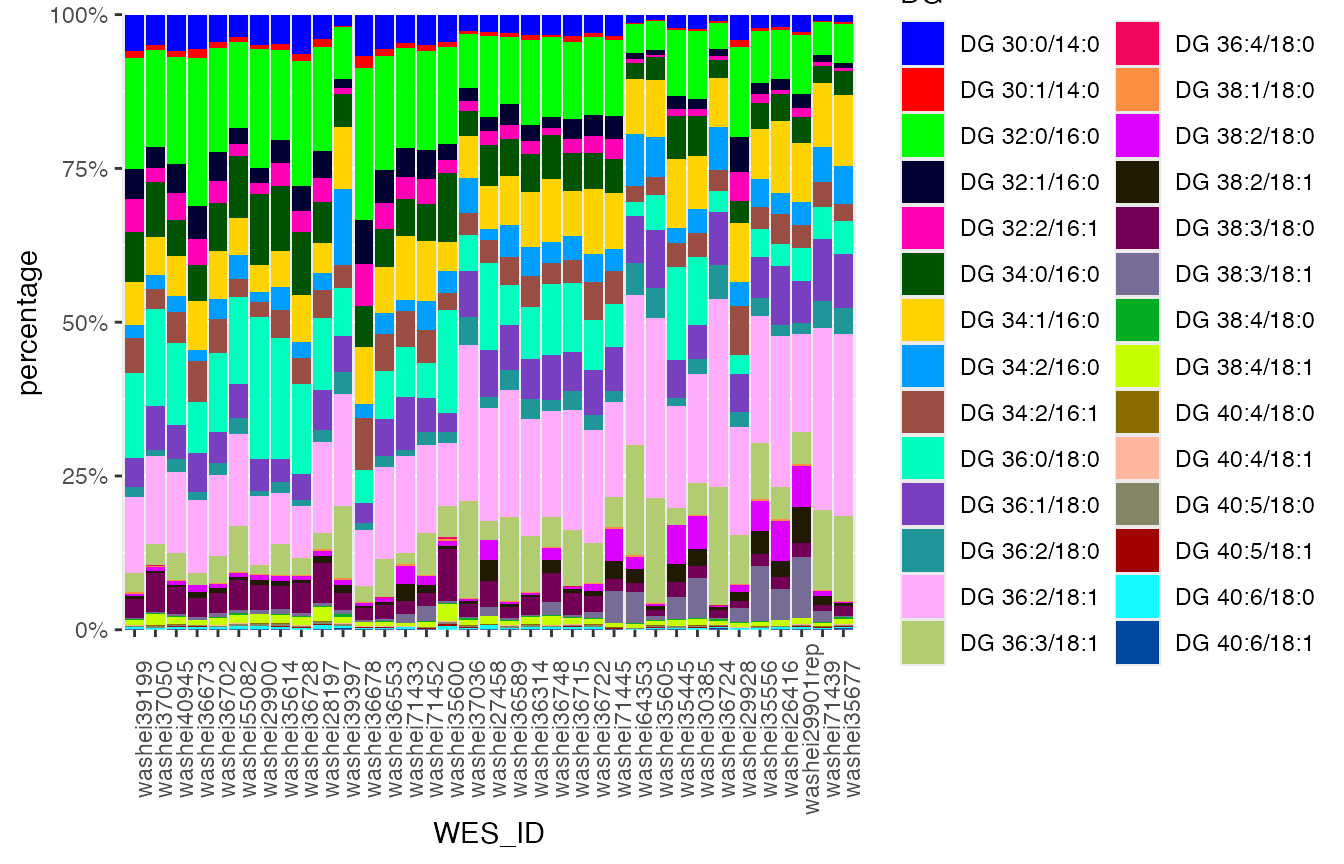
#> 15 apoB (nmol/L) %Lp(a)-ApoB100 -0.12428106 3.659771e-01Composition Component Trends
For the illustration of trends of a composition with a set of components
lipidomic_samples %>% head(3)
#> WES_ID Lp(a) (nmol/L) DG 30:0/14:0 DG 30:1/14:0 DG 32:0/16:0
#> 1 washei26416 157.0 0.0008071535 0.0001372855 0.003098725
#> 2 washei27458 37.1 0.0009333268 0.0002105400 0.004237114
#> 3 washei28197 15.0 0.0032868436 0.0010452749 0.013764988
#> DG 32:1/16:0 DG 32:2/16:1 DG 34:0/16:0 DG 34:1/16:0 DG 34:2/16:0 DG 34:2/16:1
#> 1 0.0006956733 0.0002478549 0.001698103 0.004516096 0.0013578084 0.001842253
#> 2 0.0007368318 0.0007199269 0.002174789 0.002236245 0.0005861636 0.001207711
#> 3 0.0036933192 0.0031672078 0.005407289 0.004072219 0.0021591445 0.003816118
#> DG 36:0/18:0 DG 36:1/18:0 DG 36:2/18:0 DG 36:2/18:1 DG 36:3/18:1 DG 36:4/18:0
#> 1 0.001415583 0.003683802 0.0007110398 0.009479445 0.0020066608 9.143163e-06
#> 2 0.004551087 0.002515179 0.0005592532 0.005935862 0.0009731812 2.037506e-05
#> 3 0.009499735 0.005311344 0.0016281908 0.012059293 0.0022217465 0.000000e+00
#> DG 38:1/18:0 DG 38:2/18:0 DG 38:2/18:1 DG 38:3/18:0 DG 38:3/18:1 DG 38:4/18:0
#> 1 0.0001244584 0.0025175506 0.0009816070 0.0007872274 0.0019228689 4.408206e-05
#> 2 0.0000000000 0.0010531250 0.0011219876 0.0013524833 0.0004459673 4.037663e-05
#> 3 0.0002654990 0.0006729207 0.0008393582 0.0053460809 0.0004614999 1.576657e-04
#> DG 38:4/18:1 DG 40:4/18:0 DG 40:4/18:1 DG 40:5/18:0 DG 40:5/18:1 DG 40:6/18:0
#> 1 0.0003366250 1.112712e-05 1.190158e-05 4.217323e-05 1.682002e-05 9.500192e-05
#> 2 0.0003986452 2.709289e-05 1.349291e-05 5.475041e-06 1.950699e-05 1.778880e-04
#> 3 0.0018140866 8.260741e-05 9.626816e-05 2.173361e-04 6.999751e-05 5.937550e-04
#> DG 40:6/18:1
#> 1 7.053647e-05
#> 2 5.693585e-05
#> 3 1.035781e-04You can either specify the column you want by variable = c(“col1”, “col2”, “col3”) if you need, but most of the times it will be quite lengthy. If it is not specified, it will use all the columns not mentioned in the table.
mirrorstoolkit::trend_bar_chart(
data = lipidomic_samples,
id = "WES_ID",
by = "Lp(a) (nmol/L)",
legend_title = "DG"
)
Association Studies
For those you are interested, you might want to log them if you think they are not normally distributed:
df = df %>% dplyr::mutate(
LDL = `LDL (calc) mg/dL`,
lpa_logged = `Lp(a) (nmol/L)` %>% base::log2(),
)OLS_wrapper and logistic_wrapper() can deal with most of the association analysis we need. In most of our tasks, we can specify the follow 2 or 3 lists as global settings used repetitively.
# The adjustment variable you need
adjustments = c(
"Age",
"Gender",
"AF",
"BMI",
"Smoking",
"antiplatelet_group",
"anticoagulant_group",
"statin_group"
)
# The variable you are interested in
variable_list = c(
"Albumin",
"Calcium",
"LDL",
"wIS",
"lpa_logged")
# In case the column name is not what you want in the output
variable_list_formal = c(
"Albumin",
"Calcium",
"LDL(calc) mg/dL",
"wIS",
"Lp(a) nmol/L, 2-logged"
)Logistics regression
The wrapper is runing in the following way:
- For each column name mentioned in variable_list, let’s call
it variable_i
- Grab the columns mentioned in variable_i, adjustments, and response from data. If variable_i is in adjustments, will remove the duplicates
- Logistic regression response variable_i + adjustment + 1
- Seek the effect size and coefficient of variable_i, compute the odd ratio by exp(effect size)
- Collect all the data
What you need to specify is:
- data, your dataframe
- response, which the column name of your target (y)
- adjustments, you adjustment list defined above
- by, if you want to categorize the data
- variable_of_interest, your variable list
- variable_of_interest_formal_name, the variable name list in the data
mirrorstoolkit::logistic_wrapper(
data = df,
response = "Calciphylaxis",
adjustments = adjustments,
variable_of_interest = variable_list,
variable_of_interest_formal_name = variable_list_formal
)
#> Effect Size Odd Ratio P-value
#> Albumin -5.170826684 0.005679871 0.00327561
#> Calcium 0.833885757 2.302247357 0.11224877
#> LDL(calc) mg/dL 0.006642994 1.006665108 0.61218217
#> wIS 0.003948747 1.003956554 0.95723792
#> Lp(a) nmol/L, 2-logged 0.014135081 1.014235454 0.94323132
mirrorstoolkit::logistic_wrapper(
data = df,
response = "Calciphylaxis",
adjustments = adjustments,
variable_of_interest = variable_list,
variable_of_interest_formal_name = variable_list_formal,
with_power = TRUE,
variable_distribution = c("normal", "normal", "normal", "normal", "normal")
)
#> +--------------------------------------------------+
#> | POWER CALCULATION |
#> +--------------------------------------------------+
#>
#> Logistic Regression Coefficient (Wald's Z-Test)
#>
#> Method : Demidenko (Variance Corrected)
#> Predictor Dist. : Normal
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : Odds Ratio = 1
#> H1 (Alt. Claim) : Odds Ratio != 1
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 52
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error (beta) = 0.970
#> Statistical Power = 0.03 <<
#>
#> +--------------------------------------------------+
#> | SAMPLE SIZE CALCULATION |
#> +--------------------------------------------------+
#>
#> Logistic Regression Coefficient (Wald's Z-Test)
#>
#> Method : Demidenko (Variance Corrected)
#> Predictor Dist. : Normal
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : Odds Ratio = 1
#> H1 (Alt. Claim) : Odds Ratio != 1
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 243311 <<
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error (beta) = 0.200
#> Statistical Power = 0.8
#>
#> +--------------------------------------------------+
#> | POWER CALCULATION |
#> +--------------------------------------------------+
#>
#> Logistic Regression Coefficient (Wald's Z-Test)
#>
#> Method : Demidenko (Variance Corrected)
#> Predictor Dist. : Normal
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : Odds Ratio = 1
#> H1 (Alt. Claim) : Odds Ratio != 1
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 52
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error (beta) = 0.948
#> Statistical Power = 0.052 <<
#>
#> +--------------------------------------------------+
#> | SAMPLE SIZE CALCULATION |
#> +--------------------------------------------------+
#>
#> Logistic Regression Coefficient (Wald's Z-Test)
#>
#> Method : Demidenko (Variance Corrected)
#> Predictor Dist. : Normal
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : Odds Ratio = 1
#> H1 (Alt. Claim) : Odds Ratio != 1
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 26460 <<
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error (beta) = 0.200
#> Statistical Power = 0.8
#>
#> +--------------------------------------------------+
#> | POWER CALCULATION |
#> +--------------------------------------------------+
#>
#> Logistic Regression Coefficient (Wald's Z-Test)
#>
#> Method : Demidenko (Variance Corrected)
#> Predictor Dist. : Normal
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : Odds Ratio = 1
#> H1 (Alt. Claim) : Odds Ratio != 1
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 56
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error (beta) = 0.911
#> Statistical Power = 0.089 <<
#>
#> +--------------------------------------------------+
#> | SAMPLE SIZE CALCULATION |
#> +--------------------------------------------------+
#>
#> Logistic Regression Coefficient (Wald's Z-Test)
#>
#> Method : Demidenko (Variance Corrected)
#> Predictor Dist. : Normal
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : Odds Ratio = 1
#> H1 (Alt. Claim) : Odds Ratio != 1
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 1244 <<
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error (beta) = 0.200
#> Statistical Power = 0.8
#>
#> +--------------------------------------------------+
#> | POWER CALCULATION |
#> +--------------------------------------------------+
#>
#> Logistic Regression Coefficient (Wald's Z-Test)
#>
#> Method : Demidenko (Variance Corrected)
#> Predictor Dist. : Normal
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : Odds Ratio = 1
#> H1 (Alt. Claim) : Odds Ratio != 1
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 54
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error (beta) = 0.950
#> Statistical Power = 0.05 <<
#>
#> +--------------------------------------------------+
#> | SAMPLE SIZE CALCULATION |
#> +--------------------------------------------------+
#>
#> Logistic Regression Coefficient (Wald's Z-Test)
#>
#> Method : Demidenko (Variance Corrected)
#> Predictor Dist. : Normal
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : Odds Ratio = 1
#> H1 (Alt. Claim) : Odds Ratio != 1
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 101680 <<
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error (beta) = 0.200
#> Statistical Power = 0.8
#>
#> +--------------------------------------------------+
#> | POWER CALCULATION |
#> +--------------------------------------------------+
#>
#> Logistic Regression Coefficient (Wald's Z-Test)
#>
#> Method : Demidenko (Variance Corrected)
#> Predictor Dist. : Normal
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : Odds Ratio = 1
#> H1 (Alt. Claim) : Odds Ratio != 1
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 54
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error (beta) = 0.949
#> Statistical Power = 0.051 <<
#>
#> +--------------------------------------------------+
#> | SAMPLE SIZE CALCULATION |
#> +--------------------------------------------------+
#>
#> Logistic Regression Coefficient (Wald's Z-Test)
#>
#> Method : Demidenko (Variance Corrected)
#> Predictor Dist. : Normal
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : Odds Ratio = 1
#> H1 (Alt. Claim) : Odds Ratio != 1
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 49642 <<
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error (beta) = 0.200
#> Statistical Power = 0.8
#> Effect Size Odd Ratio P-value current power
#> Albumin -5.170826684 0.005679871 0.00327561 0.03017726
#> Calcium 0.833885757 2.302247357 0.11224877 0.05158879
#> LDL(calc) mg/dL 0.006642994 1.006665108 0.61218217 0.08874263
#> wIS 0.003948747 1.003956554 0.95723792 0.05045103
#> Lp(a) nmol/L, 2-logged 0.014135081 1.014235454 0.94323132 0.05091651
#> n_observations 80%_power_size
#> Albumin 52 243311
#> Calcium 52 26460
#> LDL(calc) mg/dL 56 1244
#> wIS 54 101680
#> Lp(a) nmol/L, 2-logged 54 49642Ordinary Linear Regression
The setting is similar, let’s generate a continuous target variable:
df_disease =df %>%
dplyr::filter(Calciphylaxis == 1) %>%
dplyr::mutate(
duration_before_diagnosis = (Calciphylaxis_diagnosis_date - HD_start_date) %>%
lubridate::as.duration()/ lubridate::dmonths(1),
duration_before_diagnosis = pmax(duration_before_diagnosis,0))
df_disease$duration_before_diagnosis
#> [1] 78.7186858 47.6714579 92.9117043 39.3593429 0.0000000 2.2340862
#> [7] 23.7535934 3.9425051 26.0205339 2.9240246 23.1950719 2.9568789
#> [13] 28.3860370 0.7227926 65.5770021 9.3963039 3.7453799 83.8439425
#> [19] 27.2032854 0.0000000 24.7392197 12.4517454 10.8418891 28.4517454
#> [25] 1.9712526 50.9240246 0.9527721I called it OLS because machine learning community call it so… Forgive me. Ordinary linear regression doesn’t have OR defined thus only beta and p-value here.
mirrorstoolkit::OLS_wrapper(
data = df_disease,
response = "duration_before_diagnosis",
adjustments = adjustments,
variable_of_interest = variable_list,
variable_of_interest_formal_name = variable_list_formal
)
#> Effect Size P-value
#> Albumin 9.38512237 0.419285024
#> Calcium 23.95816412 0.004014825
#> LDL(calc) mg/dL 0.08337316 0.749217682
#> wIS -1.01676960 0.442564703
#> Lp(a) nmol/L, 2-logged 0.51243742 0.906371748
mirrorstoolkit::OLS_wrapper(
data = df_disease,
response = "duration_before_diagnosis",
adjustments = adjustments,
variable_of_interest = variable_list,
variable_of_interest_formal_name = variable_list_formal,
with_power = TRUE,
variable_distribution = c("normal","normal","normal","normal","normal")
)
#> +--------------------------------------------------+
#> | POWER CALCULATION |
#> +--------------------------------------------------+
#>
#> Linear Regression Coefficient (T-Test)
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : beta - null.beta = 0
#> H1 (Alt. Claim) : beta - null.beta != 0
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 27
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error = 0.772
#> Statistical Power = 0.228 <<
#>
#> +--------------------------------------------------+
#> | SAMPLE SIZE CALCULATION |
#> +--------------------------------------------------+
#>
#> Linear Regression Coefficient (T-Test)
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : beta - null.beta = 0
#> H1 (Alt. Claim) : beta - null.beta != 0
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 131 <<
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error = 0.198
#> Statistical Power = 0.802
#> Warning: `r.squared` is possibly larger.
#> +--------------------------------------------------+
#> | POWER CALCULATION |
#> +--------------------------------------------------+
#>
#> Linear Regression Coefficient (T-Test)
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : beta - null.beta = 0
#> H1 (Alt. Claim) : beta - null.beta != 0
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 27
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error = 0.000
#> Statistical Power = 1 <<
#> Warning: `r.squared` is possibly larger.
#> +--------------------------------------------------+
#> | SAMPLE SIZE CALCULATION |
#> +--------------------------------------------------+
#>
#> Linear Regression Coefficient (T-Test)
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : beta - null.beta = 0
#> H1 (Alt. Claim) : beta - null.beta != 0
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 14 <<
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error = 0.098
#> Statistical Power = 0.902
#>
#> +--------------------------------------------------+
#> | POWER CALCULATION |
#> +--------------------------------------------------+
#>
#> Linear Regression Coefficient (T-Test)
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : beta - null.beta = 0
#> H1 (Alt. Claim) : beta - null.beta != 0
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 27
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error = 0.922
#> Statistical Power = 0.078 <<
#>
#> +--------------------------------------------------+
#> | SAMPLE SIZE CALCULATION |
#> +--------------------------------------------------+
#>
#> Linear Regression Coefficient (T-Test)
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : beta - null.beta = 0
#> H1 (Alt. Claim) : beta - null.beta != 0
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 775 <<
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error = 0.200
#> Statistical Power = 0.8
#>
#> +--------------------------------------------------+
#> | POWER CALCULATION |
#> +--------------------------------------------------+
#>
#> Linear Regression Coefficient (T-Test)
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : beta - null.beta = 0
#> H1 (Alt. Claim) : beta - null.beta != 0
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 26
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error = 0.823
#> Statistical Power = 0.177 <<
#>
#> +--------------------------------------------------+
#> | SAMPLE SIZE CALCULATION |
#> +--------------------------------------------------+
#>
#> Linear Regression Coefficient (T-Test)
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : beta - null.beta = 0
#> H1 (Alt. Claim) : beta - null.beta != 0
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 174 <<
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error = 0.198
#> Statistical Power = 0.802
#>
#> +--------------------------------------------------+
#> | POWER CALCULATION |
#> +--------------------------------------------------+
#>
#> Linear Regression Coefficient (T-Test)
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : beta - null.beta = 0
#> H1 (Alt. Claim) : beta - null.beta != 0
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 26
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error = 0.945
#> Statistical Power = 0.055 <<
#>
#> +--------------------------------------------------+
#> | SAMPLE SIZE CALCULATION |
#> +--------------------------------------------------+
#>
#> Linear Regression Coefficient (T-Test)
#>
#> ---------------------------------------------------
#> Hypotheses
#> ---------------------------------------------------
#> H0 (Null Claim) : beta - null.beta = 0
#> H1 (Alt. Claim) : beta - null.beta != 0
#>
#> ---------------------------------------------------
#> Results
#> ---------------------------------------------------
#> Sample Size = 4078 <<
#> Type 1 Error (alpha) = 0.050
#> Type 2 Error = 0.200
#> Statistical Power = 0.8
#> Effect Size P-value current power n_observations
#> Albumin 9.38512237 0.419285024 0.22837201 27
#> Calcium 23.95816412 0.004014825 0.99992770 27
#> LDL(calc) mg/dL 0.08337316 0.749217682 0.07848074 27
#> wIS -1.01676960 0.442564703 0.17692901 26
#> Lp(a) nmol/L, 2-logged 0.51243742 0.906371748 0.05509955 26
#> 80%_power_size
#> Albumin 131
#> Calcium 14
#> LDL(calc) mg/dL 775
#> wIS 174
#> Lp(a) nmol/L, 2-logged 4078Summarize association studies
Manhattan Plot
A quick Manhattan plot API is provided. A minimal data input can be the following
association_result %>% head(3)
#> # A tibble: 3 × 4
#> SNP pos phenotype `FDR_adjusted_p-value`
#> <chr> <dbl> <chr> <dbl>
#> 1 412-C/T 412 Glucose 0.0220
#> 2 4785-C/A 4785 Hypertension 0.0316
#> 3 387-G/C 387 Hypertension 0.0316Simply provide the column names to the API.
- pos: the position of SNV
- phenotype: the phenotype it links to
- p_value: the p_value of the association
- formal_name: give name to each SNV
- phenotype_list: the list of phenotype you want to work on.
- p_value_threshold: default 0.05, which will affect the significance level (red horizontal line), all points above this line should be labelled unless there are too many overlapping
mirrorstoolkit::manhattan_plot(
data = association_result,
pos = "pos",
phenotype = "phenotype",
p_value = "FDR_adjusted_p-value",
formal_name = "SNP",
phenotype_list = c("Hypertension", "HDL", "Glucose")
) It’s also okay for single phenotype, with another p-value threshold
It’s also okay for single phenotype, with another p-value threshold
mirrorstoolkit::manhattan_plot(
data = association_result,
pos = "pos",
phenotype = "phenotype",
p_value = "FDR_adjusted_p-value",
formal_name = "SNP",
phenotype_list = "Hypertension",
p_value_threshold = 0.10
)
Directed Acyclic Graph for multi-omics data
For data like these…
multi_omics_association_result %>% head(5)
#> # A tibble: 5 × 6
#> from to beta `FDR_adjusted_p-value` from_type to_type
#> <chr> <chr> <dbl> <dbl> <chr> <chr>
#> 1 4894-G/A lpa 1.36 0.0211 genotype_SNP phenotype
#> 2 620-A/G lpa 1.43 0.0211 genotype_SNP phenotype
#> 3 621-T/A lpa 1.43 0.0211 genotype_SNP phenotype
#> 4 4922-C/G lpa 1.49 0.0211 genotype_SNP phenotype
#> 5 4970-T/A lpa 1.35 0.0211 genotype_SNP phenotypeYou can quickly generate these by networkX, it can be saved as an interactive html files if you need.
dag = mirrorstoolkit::multi_omics_directed_acyclic_graph(
data = multi_omics_association_result,
from = "from",
to = "to",
beta = "beta",
p_value = "FDR_adjusted_p-value",
from_class = "from_type",
to_class = "to_type"
)
dag$illustration_networkVisualization of restricted cubic spline model
head(df)
#> factor1 factor2 age bmi sex
#> 1 1.0685479 0.680923388 57.94493 20.83511 0
#> 2 0.9717651 1.013496371 58.20817 25.20727 0
#> 3 1.0181564 -0.009105223 53.33630 26.60876 0
#> 4 1.0316431 0.560053416 52.09588 29.94023 0
#> 5 1.0202134 -1.478541110 55.37792 30.23393 1
#> 6 0.9946938 0.952096837 51.99340 19.04435 1
head(y)
#> [1] 1 0 0 1 0 1
dd = rms::datadist(df)
lrm_fit <- rms::lrm(
y ~ rms::rcs(factor1,3)*rms::rcs(factor2,3)+ bmi+sex+age,
data = df)
mirrorstoolkit::rms_illustration_3d(
fit = lrm_fit,
datadist = dd,
variable = c("factor1", "factor2"),
target_name = "target",
density = 30)
#> Warning in formula.character(object, env = baseenv()): Using formula(x) is deprecated when x is a character vector of length > 1.
#> Consider formula(paste(x, collapse = " ")) instead.